Unlocking the Histone Code: A Spotlight on Three Essential Histone PTMs

Introduction
The “histone code” hypothesis posits that epigenetic features, specifically histone post-translational modifications (PTMs), act as a unique molecular language to regulate chromatin structure and gene expression1-3. But how does this work?
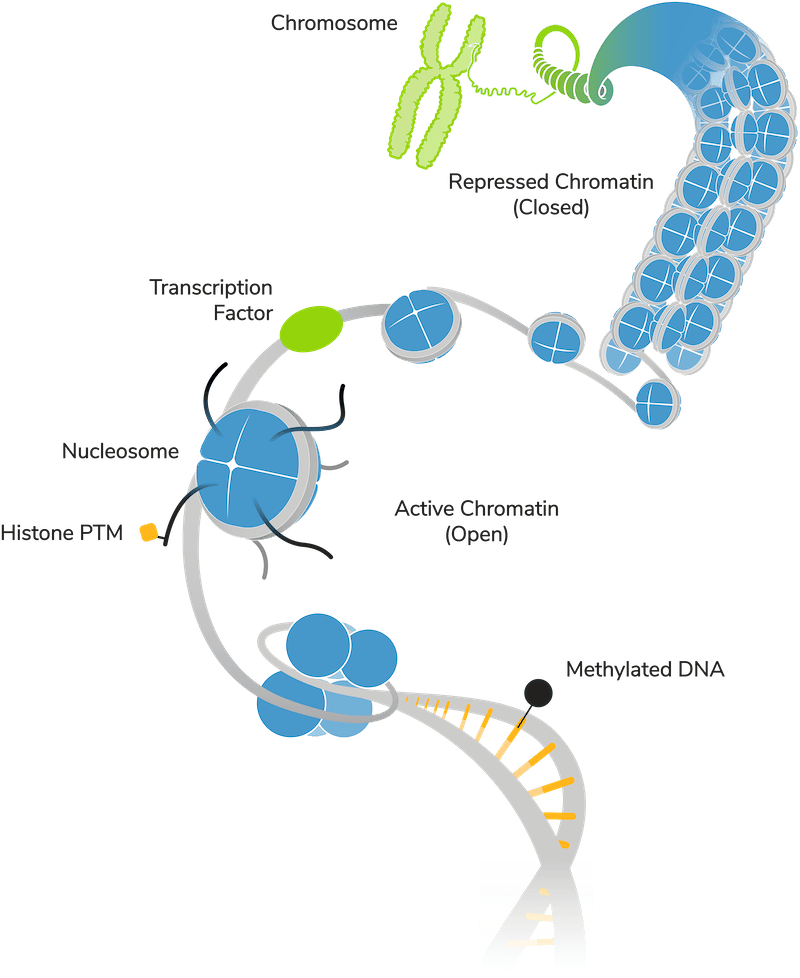
Histone PTMs occur on nucleosomes, the basic repeating unit of chromatin, composed of a histone octamer core wrapped by ~147 bp of DNA4. PTMs on the tails of histone proteins can make nucleosomes more compact or "closed”, creating dense heterochromatin to block transcriptional activation (Figure 1). Histone PTMs can also promote an “open” chromatin structure, making DNA more accessible and/or creating protein binding sites, which support gene expression5.
Different histone PTMs or combinations of histone PTMs have distinct impacts on chromatin structure. Many histone PTMs also engage in “crosstalk” with other chromatin features, such as DNA methylation, creating multi-layered systems of transcriptional control6. Understanding the biological implication of PTMs is critical to understanding how chromatin controls gene expression, and disease development, and other physiological processes.
In this article, we will dissect three widely studied histone PTM categories – acetylation, methylation, and phosphorylation – unraveling their function and how they impact on our biological makeup.
Histone Acetylation: Disruption of nucleosome contacts promotes gene activation
First on our list is histone acetylation, a highly abundant modification that is associated with gene activation. Enzymes known as histone acetyltransferases (HATs) attach an acetyl group to histone lysine residues. This process neutralizes the positive charge of histones, loosening histone-DNA interactions and (in some cases) destabilizing nucleosome structure5,7,8. Overall, this relaxing of chromatin architecture allows greater accessibility for transcriptional machinery, which promotes gene expression5,7,8.
Studies of HATs and histone deacetylation enzymes (HDACs) have demonstrated that histone acetylation generally has rapid turnover in the cell8 and plays important roles in cell division5,8.
The impact of histone lysine acetylation on chromatin structure varies, depending on the location of the lysine residue on the histone, interacting proteins, and other factors:
- Acetylation of lysine residues on histone tails, such as H3K27ac, reduces intra-nucleosome interactions, particularly between DNA and histones9. Histone tail acetylation counteracts repressive methylation marks at promoters and enhancers, supporting gene activation.
- Histone residues in the nucleosome core (e.g. H3K56, H3K64) can also be acetylated. These marks have been shown to disrupt electrostatic interactions with DNA, which can help eject nucleosomes at promoters and enhancers to support transcription5. H4K16 acetylation is specifically indicated in maintaining inter-nucleosomal binding, which is important for maintaining three-dimensional chromatin structure10,11.
- Many proteins can bind or “read” acetylated lysine, leading to recruitment of larger chromatin modifying and/or remodeling complexes, transcriptional activators, or even histone chaperone proteins5. The best studied readers of lysine acetyl marks are bromodomain-containing proteins (e.g. BRD4), YEATS domain proteins (e.g. AF9), and double plant homeodomain proteins (e.g. MOZ).
Shop our histone binding domains
Disease associations
- HDACs are overexpressed in many types of cancers12. In fact, the broad-spectrum HDAC inhibitors vorinostat and romidepsin were among the earliest epigenetic-targeted drugs approved by the FDA for cancer therapy12,13.
- Histone acetylation is also linked to memory formation, aging, and neurodegenerative diseases14,15. HDAC inhibitors have shown promising results in mouse models of Alzheimer’s disease, Huntington’s disease, and others, pointing to new avenues of drug research15,16.
Histone Methylation: Context-specific regulation of gene expression
Histone methylation, catalyzed by histone methyltransferases, involves the addition of one to three methyl groups to the lysine or arginine residues of histone proteins. Because histone lysine methylation is more widely studied, our review is focused on these marks. For a recent review of arginine methylation, see 17-19.
Unlike acetylation, methylation doesn't alter the charge of histones. Instead, it provides binding sites for proteins involved in gene regulation, and frequently engages with other histone PTMs and/or DNA methylation through so-called “crosstalk” mechanisms.
Histone methylation has been implicated in various biological processes including DNA repair, X-chromosome inactivation, and cellular differentiation5,20-22. Although histone methylation was thought to be stable for many years, it is in fact reversible – the discovery of the first histone lysine demethylase LSD1 (KDM1A) in 2004 was a major landmark in the field23. Many other lysine demethylases have been discovered in the following years, and LSD1 now represents a major target in cancer drug research24,25.
The effect of histone methylation on gene expression is context-dependent, meaning that it can either facilitate or repress gene expression depending on the site and degree of methylation.
- H3K4me3 is enriched at active transcription start sites (TSSs)5. In mammalian cells, H3K4me3 interacts with TAF3, a subunit of the TFIID transcription factor26. TFIID supports transcriptional initiation via binding to core promoter sequences, such as the TATA box, and acts as a landing site for the RNA Pol II preinitiation complex27. H3K4me3 has also been shown to recruit the SAGA histone acetylation complex, which may promote gene expression28. Despite its localization at TSSs and interactions with core transcriptional machinery, the requirement of H3K4me3 for gene expression is an ongoing topic of debate29-31. We recommend reference 31 for additional background.
- H3K9me3 denotes constitutively silenced heterochromatin, and its main function is to prevent expression of transposons and other repetitive elements. H3K9me3 also contributes to the development of lamin-associated domains, the term for dense heterochromatin regions tethered to the nuclear lamina32. Notably, H3K9me3 co-occurs with DNA methylation, and the complexes that generate these two features interact with one another to help maintain a condensed chromatin structure5.
- H3K27me3 is enriched at facultative heterochromatin, which means that it associates with repressed genes and enhancers. It is produced by the Polycomb Repressive Complex 2 (PRC2) and interacts with PRC2 to facilitate “spreading” of H3K27me3 into repressed gene bodies. Compared to H3K9me3, this mark occurs in a more permissive chromatin environment, enabling interactions with transcription factors, chromatin readers, and other proteins5.
H3K27me3 is tightly linked to developmental gene expression programs. This mark interacts with Polycomb Repressive Complex 1 (PRC1) to help silence non-lineage specific genes during differentiation22. H3K27me3 is also read by the bromo-adjacent homology (BAH) domain of BAHCC1, which in turn recruits histone deacetylases and other corepressors to maintain silencing33.
Interestingly, H3K27me3 at promoters largely overlaps with unmethylated CpG islands. Loss of DNA methylation leads to redistribution of H3K27me3 across the genome, and re-expression of PRC2 target genes, suggesting DNA methylation helps regulate PRC2 recruitment21,34,35.
Disease associations
Aberrant histone lysine methylation has been associated with various pathologies, highlighting the importance of this epigenetic marker in maintaining cellular homeostasis. A few key examples:
- The histone lysine demethylase LSD1 is overexpressed in many different types of cancers, from solid tumors (e.g. breast, colorectal, prostate) to blood cancers (e.g. acute myeloid leukemia, or AML), underscoring roles in cell differentiation, proliferation, and migration. Several LSD1 inhibitors are in clinical trials, as reviewed in references 24,25.
- Rearrangement and/or gene fusions of the H3K4 methyltransferase MLL1 are a hallmark of acute myelogenous leukemia (AML) and acute lymphoblastic leukemia (ALL)36. More recent work has shown that these fusion proteins also underlie some solid tumors, including lung, intestine, and endometrial cancers37. Researchers are developing inhibitors against some of the most common fusion proteins, with promising results in mice; see reference 37 for further information.
- Mutations in histone methyltransferases also underlie various neurodevelopmental disorders, including Kabuki syndrome, Sotos syndrome, and Wiedemann-Steiner syndrome38. Sotos syndrome is a childhood overgrowth disorder, driven by mutations in the H3K36 methyltransferase NSD1. Sotos syndrome phenocopies Tatton-Brown-Rahman syndrome (TBRS), which is caused by mutations in the DNA methyltransferase DNMT3A.
A 2019 study co-authored by EpiCypher established a mechanistic link between DNMT3A and NSD1, clarifying the similarities between these two diseases and demonstrating another route by which DNA methylation “talks” with histone modifications39. This discovery was supported by our dCypher™ platform, which uses modified nucleosomes to screen chromatin binding specificity in a physiologically relevant context. To learn more about this study and EpiCypher’s dCypher platform, see this blog.
Learn about EpiCypher dCypher™ Screening Services
Histone Phosphorylation: PTMs involved in rapid cellular responses
The last crucial histone modification we will discuss is phosphorylation. This process involves the addition of a phosphate group to the serine, threonine, or tyrosine residues of the histone proteins, and is catalyzed by kinases. In many cases, the phosphate modification occurs alongside another mark, such as histone lysine acetylation, or on variant histones, to create a distinct binding site for a protein or complex40.
Histone phosphorylation is a dynamic process that is critical for the regulation of the cell cycle, DNA repair, and apoptosis. Crosstalk with other chromatin modifications also allows histone phosphorylation to alter gene expression in response to external stimuli, as described below.
- γH2AX – referring to phosphorylation of histone variant H2AX – is a marker of double-stranded DNA breaks, and a hallmark of the DNA damage response41,42. γH2AX is an essential signal to the cell that something is wrong, and initiates the recruitment of proteins involved in the DNA damage response, such as MDC1, 53BP1, and MRN43. It is also one of the most widely used markers for double-stranded breaks in immunostaining44.
- Phosphorylation of H3S28 and/or H3.3S31 supports transcriptional responses in stimulated immune cells, such as macrophages45-47. These mechanisms can also involve other histone modifications. For instance, H3S28ph requires H3K27 acetylation to amplify gene expression in stimulated macrophages46.
- H3S10 phosphorylation is implicated in cell stress and immune responses and is widely known for its role in regulating chromosome condensation in mitosis48,49. H3S10ph shows crosstalk with H4K16ac as well, forming a binding site for double bromodomain protein BRD4 to promote transcription in vitro50. There are many other potential functions for H3S10 phosphorylation – see reference 48 to learn more.
Disease associations:
- Given its role in cell division and kinase-driven pathways, elevated histone phosphorylation is highly linked with tumor development, progression, and severity. High levels of histone phosphorylation are correlated with poorer prognosis in various cancers, including breast cancer, glioblastoma, and melanoma48.
- Histone phosphorylation, when compared to histone methylation or acetylation, is vastly understudied. As improved tools (antibodies, chromatin mapping technologies) emerge, additional roles for histone phosphorylation in disease, such as inflammatory or autoimmune diseases, will likely be discovered.
How are PTMs studied?
Given their functional importance, a variety of methods exist to study histone PTMs. Below we describe a few key strategies.
- Chromatin mapping: In these assays, histone PTM-specific antibodies are used to enrich chromatin regions associated with a PTM. DNA is purified and sequenced, providing a genome-wide map of PTM localization. High resolution histone PTM mapping can be achieved with EpiCypher’s CUTANA™ CUT&RUN or CUT&Tag assays – find the right assay for your project in this blog.
- Binding specificity assays: A recombinant protein/domain of interest is screened against libraries of modified histone peptides or nucleosomes to determine histone PTM binding specificity and/or explore chromatin-based mechanisms. Association of disease-relevant proteins with specific histone PTM(s) can reveal new targets for drug development. EpiCypher’s dCypher™ chromatin binding platform uses our collection of modified nucleosomes to accurately determine binding specificity in a physiological context. Learn all about dCypher™ platform applications here.
Conclusion
Histone post-translational modifications, including acetylation, methylation, and phosphorylation, provide an additional layer of gene regulation, playing a vital role in various biological processes and disease progression. The intricate interplay between these modifications, their reversibility, and their impact on health and disease continues to be a fertile ground for scientific exploration. Understanding these modifications at a granular level can pave the way for novel therapeutic strategies, particularly in cancer and neurodegenerative diseases.
References:
- Jenuwein T et al. Translating the histone code. Science 293, 1074-80 (2001). PubMed PMID: 11498575.
- Strahl BD et al. The language of covalent histone modifications. Nature 403, 41-5 (2000). PubMed PMID: 10638745.
- Turner BM. Histone acetylation and an epigenetic code. Bioessays 22, 836-45 (2000). https://doi.org/10.1002/1521-1878(200009)22:9<836::Aid-bies9>3.0.Co;2-x..
- Luger K et al. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251-60 (1997). PubMed PMID: 9305837.
- Talbert PB et al. The Yin and Yang of Histone Marks in Transcription. Annu Rev Genomics Hum Genet 22, 147-70 (2021). https://doi.org/10.1146/annurev-genom-120220-085159.
- Du J et al. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol 16, 519-32 (2015). https://doi.org/10.1038/nrm4043.
- Zhang T et al. The interplay of histone modifications - writers that read. EMBO Rep 16, 1467-81 (2015). https://doi.org/10.15252/embr.201540945.
- Shvedunova M et al. Modulation of cellular processes by histone and non-histone protein acetylation. Nat Rev Mol Cell Biol 23, 329-49 (2022). https://doi.org/10.1038/s41580-021-00441-y.
- Brower-Toland B et al. Specific contributions of histone tails and their acetylation to the mechanical stability of nucleosomes. J Mol Biol 346, 135-46 (2005). https://doi.org/10.1016/j.jmb.2004.11.056.
- Shogren-Knaak M et al. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844-7 (2006). https://doi.org/10.1126/science.1124000.
- Kalashnikova AA et al. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface 10, 20121022 (2013). https://doi.org/10.1098/rsif.2012.1022.
- Kim HJ et al. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res 3, 166-79 (2011). https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3056563/.
- Ganesan A et al. The timeline of epigenetic drug discovery: from reality to dreams. Clin Epigenetics 11, 174 (2019). https://doi.org/10.1186/s13148-019-0776-0.
- Berson A et al. Epigenetic Regulation in Neurodegenerative Diseases. Trends Neurosci 41, 587-98 (2018). https://doi.org/10.1016/j.tins.2018.05.005.
- Shukla S et al. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front Pharmacol 11, 537 (2020). https://doi.org/10.3389/fphar.2020.00537.
- Bondarev AD et al. Recent developments of HDAC inhibitors: Emerging indications and novel molecules. Br J Clin Pharmacol 87, 4577-97 (2021). https://doi.org/10.1111/bcp.14889.
- Bedford MT et al. Protein arginine methylation in mammals: who, what, and why. Mol Cell 33, 1-13 (2009). https://doi.org/10.1016/j.molcel.2008.12.013.
- Blanc RS et al. Arginine Methylation: The Coming of Age. Mol Cell 65, 8-24 (2017)https://doi.org/10.1016/j.molcel.2016.11.003
- Wu Q et al. Protein arginine methylation: from enigmatic functions to therapeutic targeting. Nature Reviews Drug Discovery 20, 509-30 (2021)https://doi.org/10.1038/s41573-021-00159-8
- Fang H et al. X Inactivation and Escape: Epigenetic and Structural Features. Front Cell Dev Biol 7, 219 (2019)https://doi.org/10.3389/fcell.2019.00219
- Kim JJ et al. Context-specific Polycomb mechanisms in development. Nature Reviews Genetics 23, 680-95 (2022)https://doi.org/10.1038/s41576-022-00499-0
- Blackledge NP et al. Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nature reviews Molecular cell biology 16, 643-9 (2015)https://doi.org/10.1038/nrm4067
- Shi Y et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119, 941-53 (2004)https://doi.org/10.1016/j.cell.2004.12.012
- Dong J et al. A comprehensive comparative study on LSD1 in different cancers and tumor specific LSD1 inhibitors. Eur J Med Chem 240, 114564 (2022)https://doi.org/10.1016/j.ejmech.2022.114564
- Noce B et al. LSD1 inhibitors for cancer treatment: Focus on multi-target agents and compounds in clinical trials. Front Pharmacol 14, 1120911 (2023)https://doi.org/10.3389/fphar.2023.1120911
- Lauberth SM et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152, 1021-36 (2013)https://doi.org/10.1016/j.cell.2013.01.052
- Patel AB et al. Recent insights into the structure of TFIID, its assembly, and its binding to core promoter. Curr Opin Struct Biol 61, 17-24 (2020)https://doi.org/10.1016/j.sbi.2019.10.001
- Vermeulen M et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 142, 967-80 (2010)https://doi.org/10.1016/j.cell.2010.08.020
- Murray SC et al. H3K4me3 is neither instructive for, nor informed by, transcription. bioRxiv 709014 (2019)https://doi.org/10.1101/gr.133728.111
- Wang H et al. H3K4me3 regulates RNA polymerase II promoter-proximal pause-release. Nature 615, 339-48 (2023)https://doi.org/10.1038/s41586-023-05780-8
- Howe FS et al. Is H3K4me3 instructive for transcription activation? Bioessays 39, 1-12 (2017)https://doi.org/10.1002/bies.201600095
- Manzo SG et al. Lamina-associated domains: Tethers and looseners. Curr Opin Cell Biol 74, 80-7 (2022)https://doi.org/10.1016/j.ceb.2022.01.004
- Fan H et al. BAHCC1 binds H3K27me3 via a conserved BAH module to mediate gene silencing and oncogenesis. Nature Genetics 52, 1384-96 (2020)https://doi.org/10.1038/s41588-020-00729-3
- Reddington JP et al. Redistribution of H3K27me3 upon DNA hypomethylation results in de-repression of Polycomb target genes. Genome Biology 14, R25 (2013)https://doi.org/10.1186/gb-2013-14-3-r25
- Brinkman AB et al. Sequential ChIP-bisulfite sequencing enables direct genome-scale investigation of chromatin and DNA methylation cross-talk. Genome Res 22, 1128-38 (2012)https://doi.org/10.1101/gr.133728.111
- Krivtsov AV et al. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer 7, 823-33 (2007)https://doi.org/10.1038/nrc2253
- Rao RC et al. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer 15, 334-46 (2015)https://doi.org/10.1038/nrc3929
- Fallah MS et al. Impaired Regulation of Histone Methylation and Acetylation Underlies Specific Neurodevelopmental Disorders. Front Genet 11, 613098 (2020)https://doi.org/10.3389/fgene.2020.613098
- Weinberg DN et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281-6 (2019)https://doi.org/10.1038/s41586-019-1534-3
- Sawicka A et al. Histone H3 phosphorylation – A versatile chromatin modification for different occasions. Biochimie 94, 2193-201 (2012)https://doi.org/https://doi.org/10.1016/j.biochi.2012.04.018
- Mah LJ et al. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 24, 679-86 (2010)
- Maréchal A et al. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5 (2013)https://doi.org/10.1101/cshperspect.a012716
- Kinner A et al. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res 36, 5678-94 (2008)https://doi.org/10.1093/nar/gkn550
- Nelms BE et al. In situ visualization of DNA double-strand break repair in human fibroblasts. Science 280, 590-2 (1998)https://doi.org/10.1126/science.280.5363.590
- Armache A et al. Histone H3.3 phosphorylation amplifies stimulation-induced transcription. Nature 583, 852-7 (2020)https://doi.org/10.1038/s41586-020-2533-0
- Josefowicz SZ et al. Chromatin Kinases Act on Transcription Factors and Histone Tails in Regulation of Inducible Transcription. Mol Cell 64, 347-61 (2016)https://doi.org/10.1016/j.molcel.2016.09.026
- Sawicka A et al. H3S28 phosphorylation is a hallmark of the transcriptional response to cellular stress. Genome Res 24, 1808-20 (2014)https://doi.org/10.1101/gr.176255.114
- Komar D et al. Rebelled epigenome: histone H3S10 phosphorylation and H3S10 kinases in cancer biology and therapy. Clin Epigenetics 12, 147 (2020)https://doi.org/10.1186/s13148-020-00941-2
- Duan Q et al. Phosphorylation of H3S10 blocks the access of H3K9 by specific antibodies and histone methyltransferase. Implication in regulating chromatin dynamics and epigenetic inheritance during mitosis. J Biol Chem 283, 33585-90 (2008)https://doi.org/10.1074/jbc.M803312200
- Zippo A et al. Histone Crosstalk between H3S10ph and H4K16ac Generates a Histone Code that Mediates Transcription Elongation. Cell 138, 1122-36 (2009)https://doi.org/10.1016/j.cell.2009.07.031