Deciphering the Histone Code with the dCypher™ Platform

Many nuclear processes are coordinated by the activity of chromatin associated proteins (CAPs) which add, remove, and recognize specific histone post-translational modifications (PTMs). The integration of these signals to influence downstream molecular processes, such as transcription, is commonly referred to as the histone code1. Directly measuring the effect of PTMs on protein-chromatin interactions is essential to understanding these processes, their disruption in disease, and developing epigenetic-targeted therapies.
However, defining these interactions is complicated by the diversity of PTMs and the difficulty of modeling the physiological chromatin environment in vitro. Historically, modified histone peptides have been the gold standard in the field due to their ease of use and production. However, recent work – including studies from EpiCypher scientists and collaborators – questions the accuracy of histone peptide-based observations and calls for a renewed focus on the nucleosome context in chromatin research2,3.
To address these concerns, EpiCypher developed the dCypher™ platform, which enables systematic characterization of modified nucleosome and CAP interactions. In this blog, we will explain the dCypher screening approach and offer a few recent examples from the literature that highlight the importance of using nucleosome substrates when studying the histone code.
Learn about dCypher Assay Services
The dCypher™ Approach
dCypher assays leverage the sensitivity of Alpha technology4 to perform quantitative measurements with small amounts of sample in a high-throughput, 96-well plate format. In a typical assay, a CAP of interest is screened for binding interactions against 10s-100s of site-specific modified nucleosome targets. The no-wash set up of the dCypher platform allows these assays to be easily performed in a just a few hours (Figure 1).
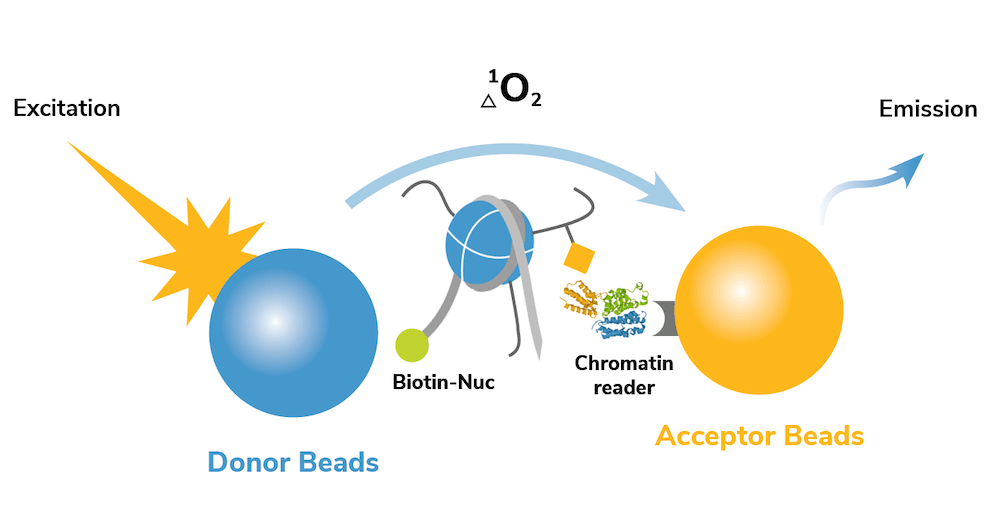
Keep reading to see how dCypher is being used by EpiCypher scientists and collaborators to drive high-impact research in chromatin biology and epigenetic drug development!
dCypher reveals novel DNMT3A-mediated cross-talk between histone PTMs and DNA methylation
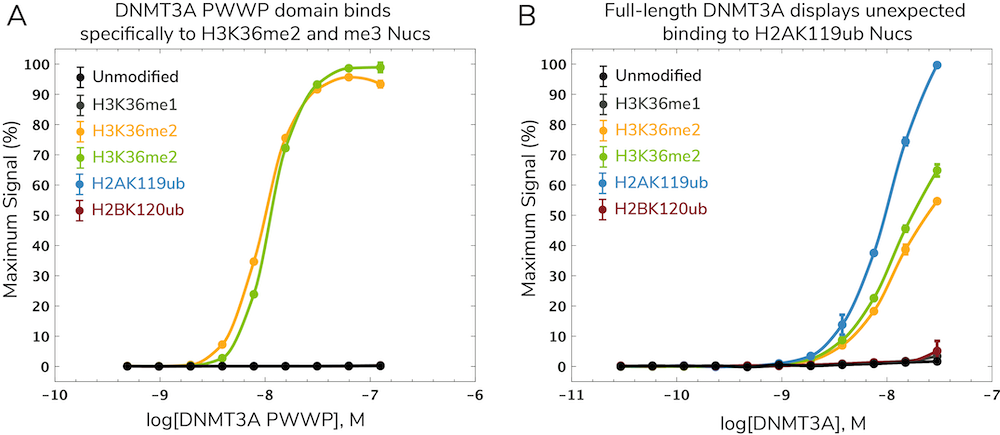
DNA methylation is an epigenetic regulatory mechanism known to interact with histone PTMs using various “cross-talk” mechanisms5. These interactions likely play roles in disease development and progression but have been challenging to study. In a recent paper, EpiCypher collaborated with the Allis lab to investigate potential interactions between the de novo DNA methyltransferase DNMT3A and the H3K36 methyltransferase NSD1, which are both associated with human overgrowth syndromes6. Specifically, we showed that the DNMT3A PWWP domain binds to H3K36me2 and me3 (Figure 2A), which helps localize DNA methylation at intergenic regions. This work was published in Nature6 with additional background on assay development described in the dCypher methods paper2. You can also check out our summary video of this work here for more context.
Although a clear relationship between H3K36me2 and intergenic DNA methylation was established, this link does not explain all instances of de novo DNA methylation. For example, DNA methylation has also been observed at CpG islands (CGIs) regulated by Polycomb repressive complexes. CGIs are not normally DNA-methylated, but they can become aberrantly methylated in disease states and repress tumor suppressor genes7.
To explore alternative recruitment mechanisms, Weinberg et al. studied full-length DNTM3A and introduced disease-associated DNMT3A "hotspot" mutations to block the PWWP-H3K36me2/me3 interaction8. If the PWWP domain is necessary for DNMT3A chromatin localization, abolishing this interaction should reduce global de novo DNA methylation.
Instead, the authors observed that mutant DNMT3A was being recruited to other regions of the genome, including Polycomb-regulated CGIs8. Weinberg et al. hypothesized that a secondary recruitment mechanism – independent of H3K36me2/me3 – could be responsible. To identify potential PTM(s) driving this novel recruitment, the authors again collaborated with EpiCypher to leverage the diversity of the dCypher platform and surveyed full-length DNMT3A interactions against modified nucleosome substrates.
Surprisingly, full-length DNMT3A showed a strong, specific interaction with ubiquitylated H2A (H2Aub) nucleosomes in addition to H3K36me2 and me3 nucleosomes (Figure 2B). H2Aub is generated by Polycomb repressive complex 1 (PRC1) activity at CGIs. This led to a model in which interactions between H2Aub and DNMT3A drive DNA methylation at CGIs, particularly when DNMT3A lacks a functional PWWP domain. These findings were corroborated by in vivo chromatin mapping data and biochemistry studies. Thus, the authors were able to identify a novel ubiquitin-dependent recruitment region of DNMT3A which contributes to mistargeting of DNMT3A and DNA methylation in human disease8.
dCypher characterizes multivalent, trans-histone interactions
PHIP (BWRD2) is a poorly characterized CAP associated with neurodevelopmental disorders and cancer. PHIP contains a Tudor domain with H3K4 methylation binding specificity9, as well as two uncharacterized bromodomains (BD1 and BD2). Bromodomains are reader domains that recognize acetylated lysines (Kac) and are often present in chromatin proteins and complexes. Histone tails can be extensively acetylated, which regulates chromatin compaction, nucleosome assembly, and transcription10.
However, it has been difficult to determine whether a histone Kac-bromodomain interaction is site-specific, as bromodomains often display promiscuous Kac binding in vitro11. Furthermore, the significant amino acid diversity of BDs makes it challenging to predict targets from primary sequence alone.
The authors teamed up with EpiCypher scientists to comprehensively investigate PHIP binding in a nucleosome context using the dCypher screening platform12. The diversity of the dCypher panels facilitated examination of each domain against a collection of Kac nucleosomes, with and without other PTMs. Using a series of point mutants that ablate binding of individual PHIP domains, the authors demonstrated that BD1 binds site-specifically to H3K14ac and BD2 binds specifically to H4K12ac.
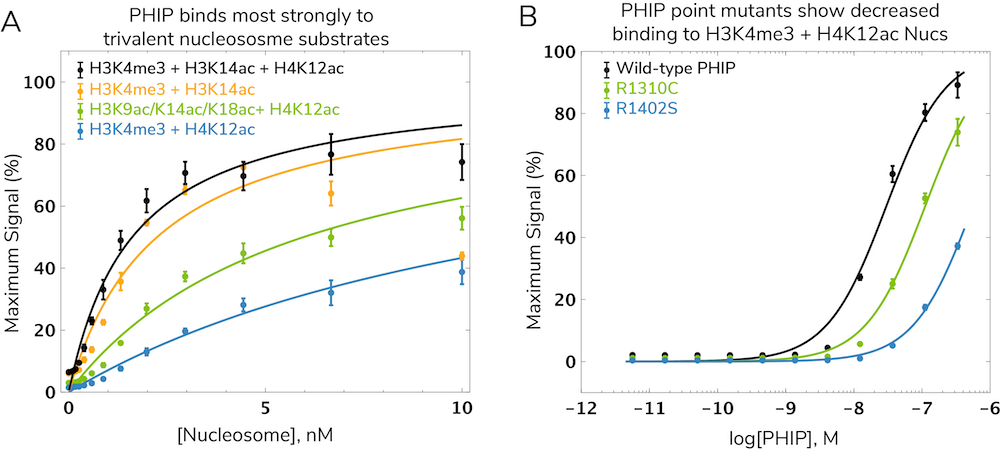
Interestingly, nucleosomes bearing all three modifications (H3K4me3, H3K14ac, and H4K12ac) show tighter binding compared to mono- or divalent nucleosomes (Figure 3A). This suggests all three marks simultaneously guide and tether PHIP to chromatin through a trans histone "cross-talk" interaction13. Evidence from PHIP proteins carrying disease-associated mutations further illustrated the requirement for all three domains to establish PHIP-nucleosome interactions12. In fact, the most prevalent PHIP mutation in human cancer (R1310C) reduced binding to H3K4me3/H4K12ac nucleosomes more than four-fold (Figure 3B).
This example underscores the importance of utilizing dCypher assays to study CAP binding in a nucleosome context, particularly when investigating the combinatorial engagement of PTMs on different histones. This study would not have been possible using modified histone peptides (the historical leading approach) or other low-throughput methods, demonstrating the power of the dCypher platform.
Getting Started – dCypher™ Nucleosome Panels and Assay Services
I have a chromatin binding protein that I would like to characterize. Can EpiCypher help? You can leverage our expertise working with CAPs and the diversity of our nucleosome substrates with our dCypher assay services. We have extensive experience characterizing CAP protein domains and full-length proteins, and the phases of our assay service have been rigorously developed to help ensure project success. This includes protein titration to determine ideal effector protein concentration, extensive buffer optimization to improve signal-to-noise and ensure specific binding, and a discovery screen to determine target binding preference.
I have a PTM (or combination of PTMs) I’m interested in studying, but it’s not included in the dCypher panels. What are my options? If we don’t have your nucleosome of interest in-house, we can make it using our custom nucleosome development services.
I want to set up the dCypher assay myself: how do I get the nucleosomes? EpiCypher offers an extensive library of highly pure modified designer nucleosomes (dNucs) and nucleosomes containing histone variants, oncogenic mutations, and methylated DNA. These nucleosomes are ready for incorporation into biochemical assays, such as binding and enzyme assays. They can also be applied to monitor antibody specificity or as assay controls. Assemble your own panel by purchasing individual nucleosomes or source one of our pre-made dCypher nucleosome panels for larger projects and greater diversity.
Is there a protocol? EpiCypher scientists recently published a dCypher protocol chapter, which provides helpful background and technical information for implementing the dCypher assay in your lab2. We also suggest reviewing our previous papers citing dCypher technology, including the ones highlighted above6,8,12,14,15.
Do you sell Alpha beads? Although dCypher uses Alpha-bead technology, the beads are purchased through Perkin-Elmer. Perkin-Elmer offers several donor/acceptor beads in addition to the ones used for dCypher, and the option to conjugate your own beads.
Explore more resources such as our dCypher product flyer and technical note to see how EpiCypher can help you decipher the histone code. Contact us at info@epicypher.com for more information!
References
- Strahl BD et al. The language of covalent histone modifications. Nature 403, 41-5 (2000). PubMed PMID: 10638745.
- Marunde MR et al. The dCypher Approach to Interrogate Chromatin Reader Activity Against Posttranslational Modification-Defined Histone Peptides and Nucleosomes. Methods Mol Biol 2458, 231-55 (2022). PubMed PMID: 35103971.
- Ghoneim M et al. Histone Tail Conformations: A Fuzzy Affair with DNA. Trends Biochem Sci (2021). PubMed PMID: 33551235.
- Ullman EF et al. Luminescent oxygen channeling immunoassay: measurement of particle binding kinetics by chemiluminescence. Proc Natl Acad Sci U S A 91, 5426-30 (1994). PubMed PMID: 8202502.
- Cedar H et al. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet 10, 295-304 (2009). PubMed PMID: 19308066.
- Weinberg DN et al. The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape. Nature 573, 281-6 (2019). PubMed PMID: 31485078.
- Deaton AM et al. CpG islands and the regulation of transcription. Genes Dev 25, 1010-22 (2011). PubMed PMID: 21576262.
- Weinberg DN et al. Two competing mechanisms of DNMT3A recruitment regulate the dynamics of de novo DNA methylation at PRC1-targeted CpG islands. Nature Genetics 53, 794-800 (2021). PubMed PMID: 33986537.
- Morgan MAJ et al. A cryptic Tudor domain links BRWD2/PHIP to COMPASS-mediated histone H3K4 methylation. Genes Dev 31, 2003-14 (2017). PubMed PMID: 29089422.
- Shahbazian MD et al. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem 76, 75-100 (2007). PubMed PMID: 17362198.
- Filippakopoulos P et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149, 214-31 (2012). PubMed PMID: 22464331.
- Morgan MAJ et al. A trivalent nucleosome interaction by PHIP/BRWD2 is disrupted in neurodevelopmental disorders and cancer. Genes Dev 35, 1642-56 (2021). PubMed PMID: 34819353.
- McGinty RK et al. Chemically ubiquitylated histone H2B stimulates hDot1L-mediated intranucleosomal methylation. Nature 453, 812-6 (2008). PubMed PMID: 18449190.
- Lloyd JT et al. Structural Insights into the Recognition of Mono- and Diacetylated Histones by the ATAD2B Bromodomain. J Med Chem 63, 12799-813 (2020). PubMed PMID: 33084328.
- Jain K et al. Characterization of the plant homeodomain (PHD) reader family for their histone tail interactions. Epigenetics Chromatin 13, 3 (2020). PubMed PMID: 31980037.