ChIP-seq vs. CUT&RUN vs. CUT&Tag: Which should you use?
- Ellen Weinzapfel

Our chromatin-mapping basics series focuses on ChIP-seq, CUT&RUN, and CUT&Tag. Each blog reviews the protocol, considerations for experimental design, and key optimization steps. But how do you decide which strategy is best for your experiment? Here we will provide guidance on how to select the best assay based on EpiCypher’s experiences in the lab.
1: Don’t use ChIP-seq
Our ChIP-seq blog thoroughly reviews the disadvantages of ChIP-seq (Figure 1), including:
- Requires millions of cells – Can’t profile rare cell types or clinical samples, limits the number of targets you can profile
- Several technically challenging steps that require optimization – Cross-linking, chromatin fragmentation, and immunoprecipitation (IP)
Note: All these steps introduce variability and sequencing artifacts, making ChIP-seq inherently noisy
- Time consuming protocol – Takes ~ 1 week to perform ChIP-seq experiment (cells to loading on sequencer), even with an optimized protocol
- Requires high sequencing depths – ChIP-seq typically requires 20-40 million reads per library for sufficient signal over background
- Poor data quality and reliability – High background and low and/or variable yields
- Low throughput – Time-intensive protocol combined with high sequencing costs and cell input requirements limit ChIP-seq to small-scale applications
Despite these challenges, ChIP-seq was the best chromatin mapping technology available for decades, and is widely used in the field. However, newer mapping strategies, including CUTANA™ CUT&RUN and CUT&Tag assays, address these problems and provide an attractive alternative to ChIP-seq (Figure 1).

CUT&Tag and CUT&RUN share many advantages in comparison to ChIP-seq. Both assays selectively target antibody-bound chromatin in intact nuclei or cells, without cross-linking, fragmentation, or IP, providing high quality profiles with low background and exquisite reliability. These same advantages also make CUTANA CUT&RUN and CUT&Tag strategies much faster than ChIP-seq and enables the use of lower cell inputs and reduced sequencing depths.
Are you sure I shouldn’t use ChIP-seq? Q&A
Scientific methods are continually evolving. This is particularly true in the epigenomics space, which has seen rapid technological growth and expansion over the past decade1-4. Despite the clear advantages of CUTANA™ assays, many researchers are hesitant to make the switch from ChIP-seq.
Adherence to outdated methods when improved strategies are readily available can delay publication and limit the impact of your research. Here we will address the most common concerns from scientists that transition from ChIP-seq to CUTANA™ assays.
I’m studying a transiently interacting protein, which requires cross-linking to stabilize target localization on chromatin. Isn’t ChIP-seq my best option?
CUT&RUN works for every target class we have tested – including transcription factors – typically under native conditions (see Figure 3 below). This allows you to generate clean data, free from the high background, sequencing artifacts, and variable IP efficiency associated with heavy cross-linking.
If desired, CUTANA assays are compatible with light to moderate cross-linking conditions (Figure 2). However, the heavy fixation strategies required for ChIP-seq should NOT be applied in CUT&RUN (or CUT&Tag).
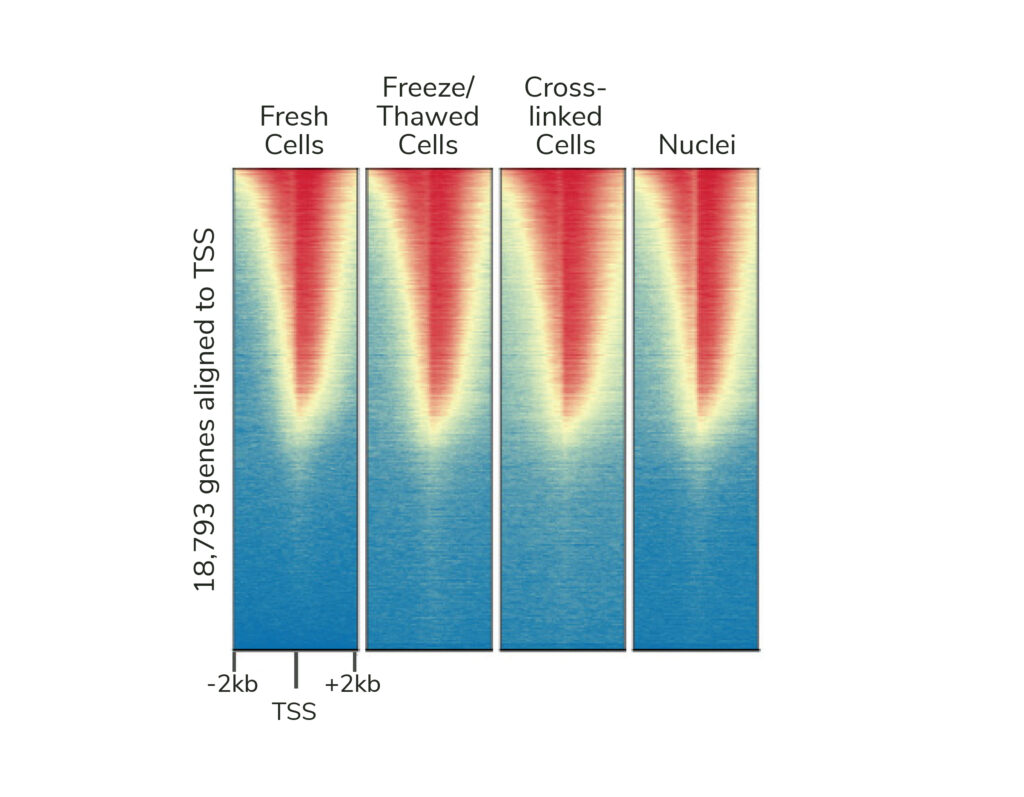
I’m trying to compare my results to existing ChIP-seq data – I need to continue doing ChIP-seq, right?
Although ChIP-seq and CUT&RUN are distinct protocols, the raw sequencing data are similar, and are processed and visualized using the same tools. Comparing these data sets is straightforward and has been published multiple times in peer-reviewed studies5-8. Furthermore, analysis of ChIP-seq and CUT&RUN data from the same cell line and target show high concordance8. The main difference is that CUT&RUN data have much lower background and require 10-fold fewer cells and sequencing reads.
I already have a good ChIP-seq protocol and/or antibody that works – shouldn’t I stick with it?
Even the best ChIP-seq protocols require more time, cells, and sequencing compared to CUT&RUN. In addition, ChIP-seq is inherently low-throughput, expensive, and has high background – problems that are fully addressed by our CUTANA™ CUT&RUN assay workflow. CUT&RUN also requires minimal optimization for most targets and cell types, in stark contrast to the labor-intensive development of target and tissue-specific cross-linking, fragmentation, and IP conditions for ChIP-seq.
What about my antibody? It works so well in ChIP!
Antibody performance is not a good justification for selecting ChIP-seq. ChIP-grade antibodies are notoriously unreliable, especially for histone PTMs9. EpiCypher found that over 70% of antibodies to histone lysine methylation and acylation PTMs display unacceptable cross-reactivity and/or target efficiency (chromatinantibodies.com). This includes highly cited antibodies for H3K4me3, H3K9me3, H3K27ac, and H3K27me3 – some of the most well-studied marks in chromatin biology. Non-histone PTM targets, such as transcription factors, face similar challenges.
Note: EpiCypher has begun validating antibodies specifically for CUT&RUN and CUT&Tag applications. Our CUT&RUN antibody collection includes reagents targeting histone PTMs, transcription factors, chromatin remodeling enzymes, reader proteins, writer enzymes, and more. Browse our CUT&RUN and CUT&Tag antibodies and subscribe at the end of this blog for updates!
You can’t sequence input samples in CUT&RUN (or CUT&Tag). How will I determine enrichment and/or examine nonspecific background?
We recommend running a negative control reaction using an antibody to IgG. IgG is an excellent control for monitoring background and/or nonspecific signal in open chromatin.
To call peaks and determine enrichment, EpiCypher routinely uses MACS210 and SICER11, peak calling programs for ChIP-seq that work well for CUT&RUN12. SICER can be adjusted for analysis of sharp enrichment peaks (e.g. H3K4me3) vs. broad areas of enrichment (e.g. H3K27me3)13. Other options include SEACR14, a peak caller designed for CUT&RUN data, and the CUT&RUNTools 2.0 pipeline, which is designed for CUT&RUN and CUT&Tag data, including analysis of single cells15. It is recommended to test several programs and select the one that faithfully represents the target of interest. See the CUTANA™ CUT&RUN Kit Manual (FAQs section) for more information.
Key Point 2: Consider CUT&RUN the “all-purpose” chromatin mapping assay
CUT&RUN is the ideal assay for most epigenomic mapping experiments. It provides a good balance between cell requirements, target compatibility (Figure 3), throughput, and sequencing costs. The protocol is simple enough to be adapted in both new and expert-level labs and is made even easier with the development of the CUTANA™ CUT&RUN Kit and the CUT&RUN Library Prep Kit.
Below we outline the advantages of CUT&RUN vs. ChIP-seq, step-by-step. We also include comparisons with CUT&Tag where appropriate.
- High-resolution data for diverse targets: CUT&RUN is compatible with histone PTMs and chromatin-associated proteins, including transcription factors, epigenetic readers, writers, and erasers (Figure 3). CUT&RUN also generates robust profiles for chromatin remodeling enzymes, which have been difficult to profile using ChIP-seq – underscoring another key advantage of CUT&RUN.

- Low cell number requirements: While it is recommended to start with 500,000 cells, CUTANA CUT&RUN can generate high quality data down to 5,000 cells with no changes to the standard protocol, enabling analysis of more targets, rare cell types, and precious samples. CUT&RUN has been used to profile mouse and human primary cells16-20, patient-derived xenografts18,21, FACS-sorted cells20,22, immune cells23-26, and more.
- Streamlined protocol: The CUTANA CUT&RUN Protocol allows users to go from starting cells to loading libraries on a sequencer in just 3 days. The protocol is also designed for use with multi-channel pipettors and 8-strip tubes, improving assay reproducibility and increasing throughput.
Note: CUT&Tag is a slightly faster assay, which may be an advantage in high-throughput applications. However, both CUT&RUN and CUT&Tag are automation-compatible18,27.
- Reduced sequencing costs: Only 3-8 million sequencing reads are required for high quality profiles, allowing more samples to be multiplexed per sequencing run.
- User-friendly workflow with less optimization: As discussed above, CUT&RUN skips the most challenging parts of ChIP-seq (chromatin fragmentation, etc.) and requires less optimization. EpiCypher has made this process even easier with our CUTANA™ CUT&RUN Kit and CUT&RUN Library Prep Kits, providing an end-to-end solution for high quality chromatin mapping.
Note: In EpiCypher’s experience, CUT&RUN is easier to learn and troubleshoot compared to CUT&Tag, particularly when using our CUT&RUN assay kits and Library Prep kits. See more on this topic below.
Key Point 3: Consider CUT&Tag the “expert-level” chromatin mapping assay
CUT&Tag is best suited for scientists with broad technical expertise in chromatin mapping assays. CUT&Tag is NOT recommended if you are:
- New to epigenomic mapping assays
- A frequent ChIP-seq user trying out CUTANA chromatin mapping assays
- Trying to map a new target and/or use a new cell type
- Mapping low abundance targets or transcription factors and other chromatin-associated proteins
In each of these scenarios, EpiCypher suggests using CUT&RUN, which has a user-friendly protocol and generates reliable profiles for most targets and cell types. Below we describe relevant drawbacks of CUT&Tag, as well as ideal applications that highlight the core advantages of this exciting technology.
CUT&Tag is more technically challenging vs. CUT&RUN
Many researchers are excited to try CUTANA CUT&Tag for their chromatin mapping experiments, because the method skips traditional library prep steps and requires only 100,000 nuclei for high-quality sequencing tracks. EpiCypher has further streamlined the CUT&Tag process with our exclusive Direct-to-PCR strategy, which allows users to go from cells to PCR amplified libraries in one tube5,28.
However, in EpiCypher’s experience, CUT&Tag assays require more practiced hands to generate robust chromatin profiles. The reduced cell input makes CUT&Tag highly sensitive to errors in assay setup and/or sample prep, ConA bead loss, and nonspecific antibodies. In addition, CUT&Tag often requires optimization and thorough knowledge of troubleshooting tactics.
For example, a common problem reported by scientists using the CUTANA™ Direct-to-PCR CUT&Tag Protocol is low or no yields following indexing PCR. Troubleshooting these concerns is far from straightforward, as there are many potential causes for reduced yields:
- Too many nuclei (can inhibit indexing PCR)
- Poor sample prep and/or too few nuclei
- Sample loss due to ConA bead dry-out
- Low abundance target, such as a transcription factor
- Poor antibody specificity and/or efficiency
CUT&Tag is also prone to higher read duplication rates (vs. CUT&RUN) and may exhibit background signal in open chromatin regions. For these reasons, we recommend CUT&RUN for most users.
CUT&Tag is not for all targets
Currently, CUTANA™ CUT&Tag is only recommended for mapping histone PTMs (Figure 4). EpiCypher does NOT recommend using CUT&Tag to map chromatin-associated proteins, which often are often weakly bound to chromatin and stripped during high-salt CUT&Tag washes. This is a major drawback of the assay, and one of the reasons we continue to suggest CUT&RUN for most users.
Note: In ChIP, samples are cross-linked to stabilize proteins on chromatin, which allows the use of stringent high-salt wash buffers. Although CUT&Tag is compatible with light to moderate cross-linking, these conditions severely reduce assay yields. Instead, EpiCypher recommends mapping protein targets in CUT&RUN using native samples.
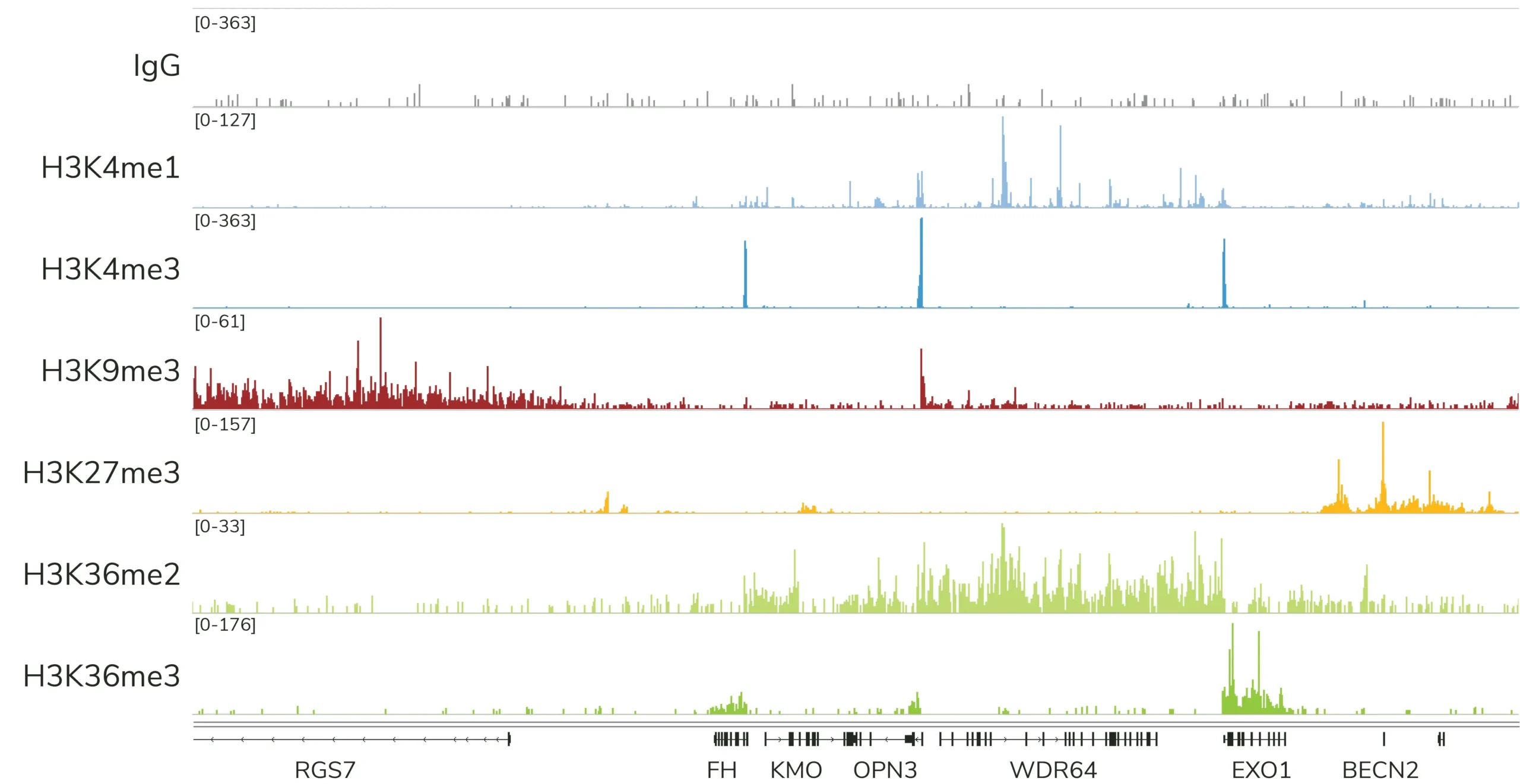
CUT&Tag is ideal for specialized histone PTM mapping applications
Many customized and/or low cell input projects are well-suited for CUT&Tag. CUT&Tag was purposely designed for chromatin mapping from small numbers of cells, providing a complementary technology to CUT&RUN5,6. The original paper tested its application for single cell profiling6, and additional modifications to the technique have increased the sensitivity of CUT&Tag for single cell epigenomics27,29-35.
Why is CUTANA™ CUT&Tag good for innovative low-input applications?
- Tn5 tagmentation eliminates traditional cross-linking, chromatin fragmentation, IP, and library prep steps, reducing hands-on time and maximizing target recovery. When trying to map from ultra-low or single cells, it is crucial to minimize processing steps. In CUT&Tag, pAG-Tn5 inserts sequencing adapters at antibody-bound chromatin in intact nuclei, removing the most time-consuming steps of ChIP-seq.
- EpiCypher’s exclusive Direct-to-PCR CUT&Tag protocol streamlines this process by allowing you to go from cells to PCR amplified DNA libraries in one tube5,28. Each time cells/DNA are washed, transferred to a new tube, or purified between steps, you risk losing material. Our unique approach requires a single DNA purification step and can be completed in just two days.
- Enables complex and/or custom multiplexing strategies, such as combinatorial indexing29-32. These approaches increase the number of individual cells that can be uniquely barcoded in a single experiment, which is key for single cell profiling. EpiCypher offers an uncharged pAG-Tn5 for researchers using or developing new CUT&Tag barcoding strategies.
Note that EpiCypher’s CUTANA CUT&Tag protocol is most robust when using 10,000 to 100,000 nuclei. We recommend using 100,000 nuclei per reaction, which will help ensure robust profiles for most histone PTMs, including low-abundance targets (i.e. H3K4me3) and PTMs in constitutive heterochromatin (i.e. H3K9me3), as shown in Figure 4. Success below 10,000 nuclei depends on multiple factors, including quality of sample prep, target abundance in your cell type, as well as antibody specificity and efficiency. Although single cell CUT&Tag data have been published using CUTANA pAG-Tn533, our current CUT&Tag protocol is not validated for single cell mapping.

Choose your chromatin mapping assay
Here is a quick checklist for helping you pick the best assay for your project:
- Don’t use ChIP-seq
- Consider CUT&RUN the go-to chromatin mapping assay, flexible for many targets, cell types, and cell processing conditions. Use CUT&RUN if you can harvest 5,000 to 500,000 cells per reaction and are:
- New to chromatin mapping and/or CUTANA™ technologies
- Mapping a new target and/or using a new cell type
- CUT&Tag is the expert-level assay for novel and/or low cell applications. It is NOT ideal for beginners. Note:
- CUT&Tag is strictly recommended for profiling histone PTMs
- These experiments often require more optimization than CUT&RUN
- The CUTANA CUT&Tag Protocol requires 10,000 to 100,000 cells per reaction for reliable PTM profiling
- Success with lower inputs is possible, but depends on multiple factors (target abundance, cell type, antibody). Genomics expertise is recommended; consult literature for guidance27,29-35.
If you still need help deciding which assay is right for you, drop us a line at [email protected].
References
- Preissl S et al. Characterizing cis-regulatory elements using single-cell epigenomics. Nat Rev Genet (2022). PubMed PMID: 35840754.
- Mehrmohamadi M et al. A Comparative Overview of Epigenomic Profiling Methods. Front Cell Dev Biol 9, 714687 (2021). PubMed PMID: 34368164.
- Carter B et al. The epigenetic basis of cellular heterogeneity. Nat Rev Genet 22, 235-50 (2021 PubMed PMID: 33244170.
- Agbleke AA et al. Advances in Chromatin and Chromosome Research: Perspectives from Multiple Fields. Mol Cell 79, 881-901 (2020). PubMed PMID: 32768408.
- Kaya-Okur HS et al. Efficient low-cost chromatin profiling with CUT&Tag. Nat Protoc 15, 3264-83 (2020). PubMed PMID: 32913232.
- Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). PubMed PMID: 31036827.
- Skene PJ et al. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat Protoc 13, 1006-19 (2018). PubMed PMID: 29651053 .
- Skene PJ et al. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, (2017). PubMed PMID: 28079019 .
- Shah RN et al. Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies. Mol Cell 72, 162-77 e7 (2018). PubMed PMID: 30244833.
- Liu T. Use model-based Analysis of ChIP-Seq (MACS) to analyze short reads generated by sequencing protein-DNA interactions in embryonic stem cells. Methods Mol Biol 1150, 81-95 (2014). PubMed PMID: 24743991.
- Zang C et al. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25, 1952-8 (2009). PubMed PMID: 19505939.
- Evans MK et al. Ybx1 fine-tunes PRC2 activities to control embryonic brain development. Nat Commun 11, 4060 (2020). PubMed PMID: 32792512.
- Laczik M et al. Iterative Fragmentation Improves the Detection of ChIP-seq Peaks for Inactive Histone Marks. Bioinform Biol Insights 10, 209-24 (2016). PubMed PMID: 27812282.
- Meers MP et al. Peak calling by Sparse Enrichment Analysis for CUT&RUN chromatin profiling. Epigenetics Chromatin 12, 42 (2019). PubMed PMID: 31300027.
- Yu F et al. CUT&RUNTools 2.0: A pipeline for single-cell and bulk-level CUT&RUN and CUT&Tag data analysis. Bioinformatics (2021). PubMed PMID: 34244724.
- Liu N et al. Direct Promoter Repression by BCL11A Controls the Fetal to Adult Hemoglobin Switch. Cell 173, 430-42 e17 (2018). PubMed PMID: 29606353.
- de Bock CE et al. HOXA9 Cooperates with Activated JAK/STAT Signaling to Drive Leukemia Development. Cancer Discov 8, 616-31 (2018). PubMed PMID: 29496663.
- Janssens DH et al. Automated in situ chromatin profiling efficiently resolves cell types and gene regulatory programs. Epigenetics Chromatin 11, 74 (2018). PubMed PMID: 30577869.
- Uyehara CM et al. Direct and widespread role for the nuclear receptor EcR in mediating the response to ecdysone in Drosophila. Proc Natl Acad Sci U S A 116, 9893-902 (2019). PubMed PMID: 31019084.
- Zhang XL et al. Reorganization of postmitotic neuronal chromatin accessibility for maturation of serotonergic identity. Elife 11, (2022). PubMed PMID: 35471146.
- Wang J et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat Cell Biol 24, 384-99 (2022). PubMed PMID: 35210568.
- Hainer SJ et al. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 177, 1319-29 e11 (2019). PubMed PMID: 30955888.
- Mathsyaraja H et al. Max deletion destabilizes MYC protein and abrogates Emicro-Myc lymphomagenesis. Genes Dev 33, 1252-64 (2019). PubMed PMID: 31395740.
- Roth TL et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 559, 405-9 (2018). PubMed PMID: 29995861.
- Collins PL et al. DNA double-strand breaks induce H2Ax phosphorylation domains in a contact-dependent manner. Nat Commun 11, 3158 (2020). PubMed PMID: 32572033.
- Yusufova N et al. Histone H1 loss drives lymphoma by disrupting 3D chromatin architecture. Nature 589, 299-305 (2021). PubMed PMID: 33299181.
- Janssens DH et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat Genet 53, 1586-96 (2021). PubMed PMID: 34663924.
- Henikoff S et al. Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. Elife 9, (2020). PubMed PMID: 33191916.
- Deng Y et al. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 375, 681-6 (2022). PubMed PMID: 35143307.
- Gopalan S et al. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol Cell 81, 4736-46 e5 (2021). PubMed PMID: 34637755.
- Xiong H et al. Single-cell joint detection of chromatin occupancy and transcriptome enables higher-dimensional epigenomic reconstructions. Nat Methods 18, 652-60 (2021). PubMed PMID: 33958790 .
- Zhu C et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods 18, 283-92 (2021). PubMed PMID: 33589836.
- Janssens DH et al. CUT&Tag2for1: a modified method for simultaneous profiling of the accessible and silenced regulome in single cells. Genome Biol 23, 81 (2022). PubMed PMID: 35300717.
- Wu SJ et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat Biotechnol (2021). PubMed PMID: 33846646.
- Bartosovic M et al. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat Biotechnol (2021). PubMed PMID: 33846645.