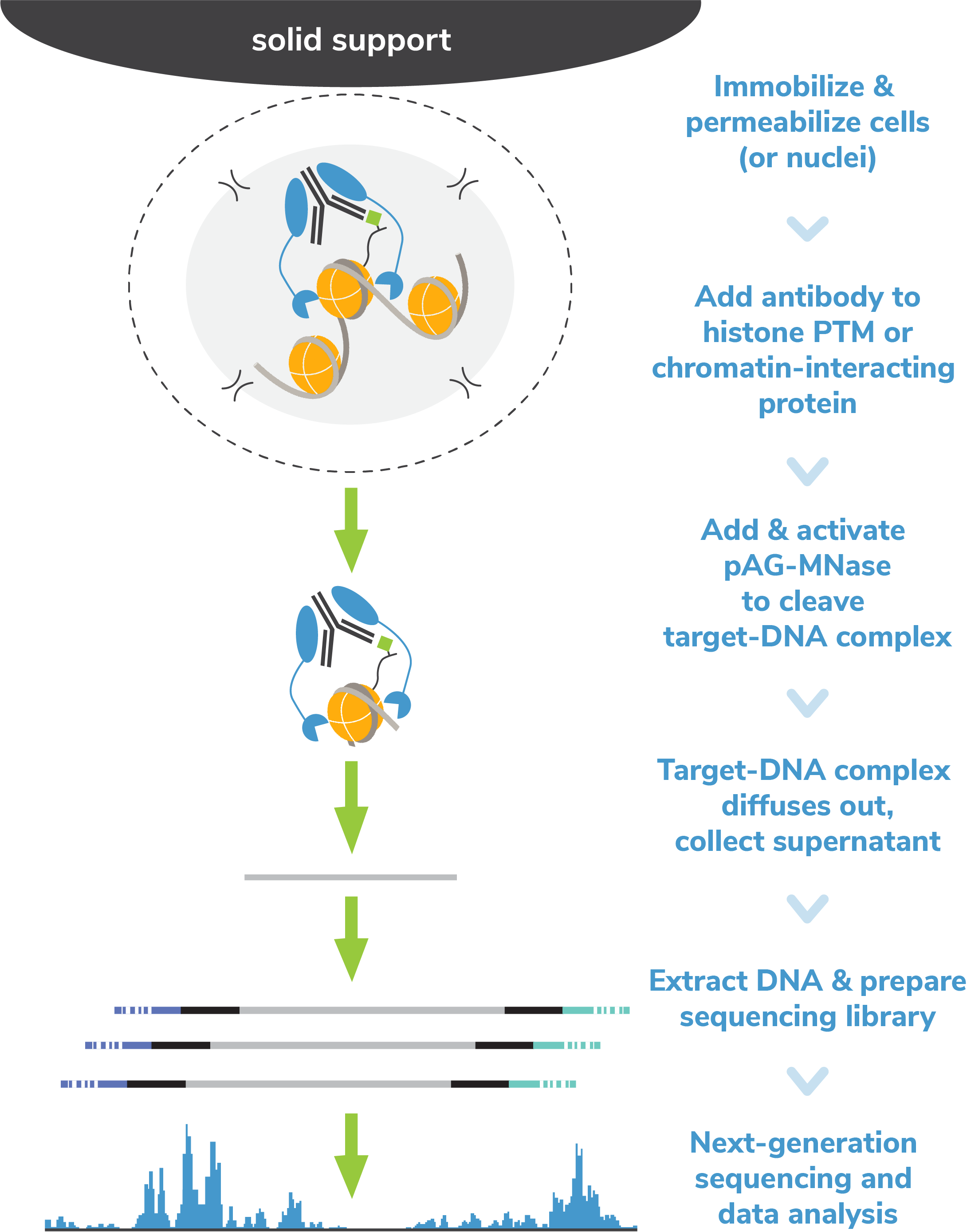
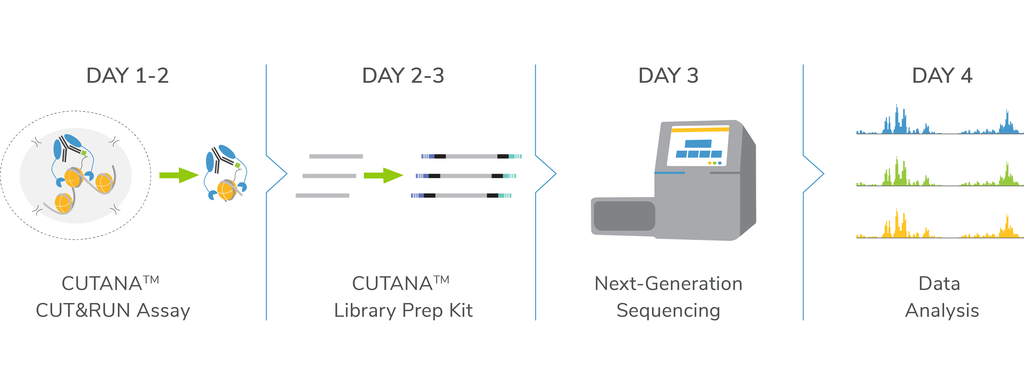

CUTANA™ CUT&RUN Kit & pAG-MNase
Jumpstart your chromatin profiling studies with our user-friendly CUT&RUN Kit or customize your workflow using pAG-MNase and our DIY protocol. For robust sequencing data, add the CUT&RUN Library Prep Kit.

CUTANA™ CUT&RUN Antibodies
EpiCypher’s CUTANA™ Antibodies are rigorously tested for reliable and robust performance in CUT&RUN assays. Check out our available chromatin-associated protein and histone PTM antibodies.

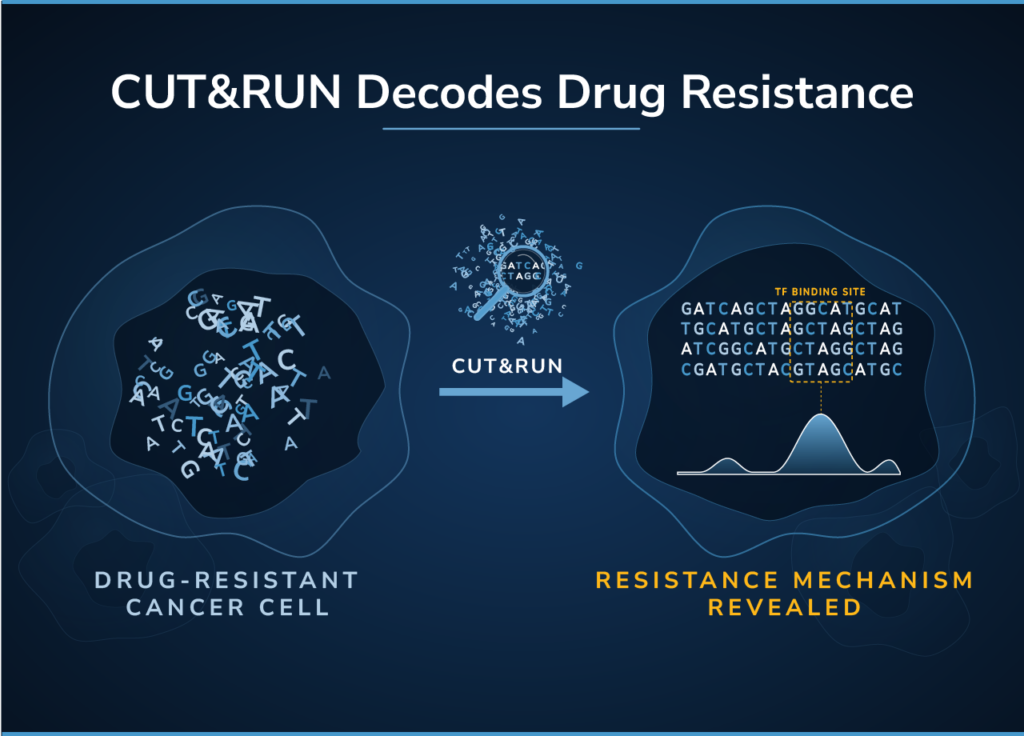
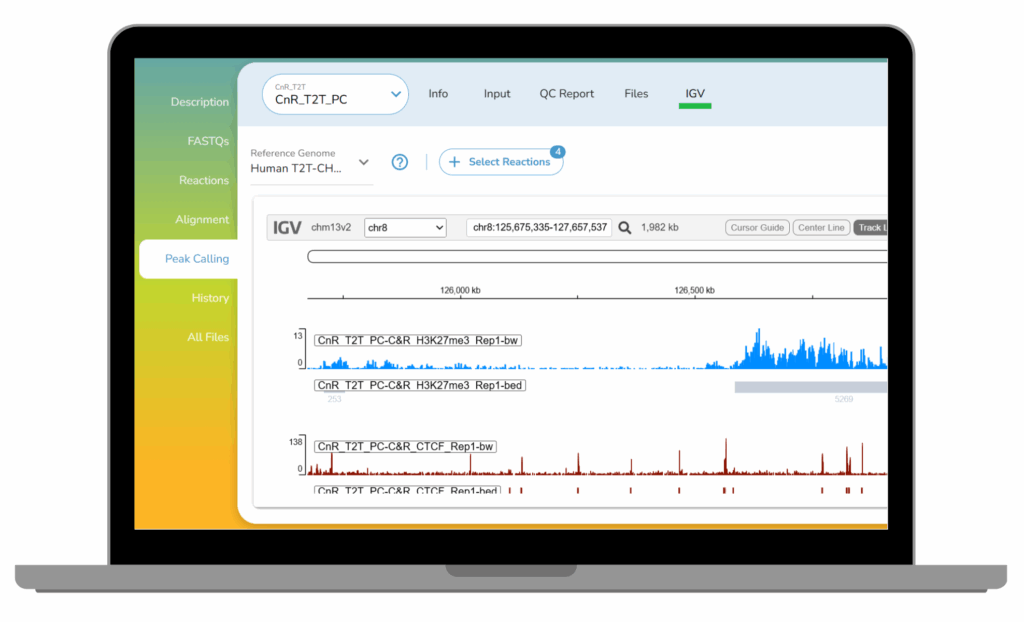
CUTANA™ CUT&RUN Services
Our CUT&RUN Services provide unique access to our genomics experts, automated CUT&RUN workflows, and in-house bioinformatic pipelines. Perfect for pilot experiments and high-throughput studies alike.