Fiber-seq 101: A Multiomic Assay That Goes Beyond ATAC-seq
- Aaron Alcala, PhD

How do cells control when and where genes are expressed? The answer lies in a complex interplay of regulatory proteins that govern chromatin accessibility and architecture. These processes establish cell identity and maintain genome stability, and when disrupted they can drive a wide range of diseases. New long-read sequencing methods such as Fiber-seq now make it possible to study chromatin with much higher sensitivity than ever before.
Weaving together a complete picture of chromatin regulation requires methods that can capture multiple chromatin features at once and at high resolution. Current approaches fall short, often requiring the integration of several different assays that slow research and use up valuable resources. Many of these assays also rely on short-read sequencing, which misses important insights in complex regions of the genome frequently tied to disease. Even widely used methods like ATAC-seq provide only a rough view of chromatin accessibility, and while this method has been shown to footprint proteins, doing so requires such deep sequencing that it is impractical for most studies.
CUTANA™ Fiber-seq is a groundbreaking multiomic approach that overcomes these challenges. By combining a simple labeling strategy with long-read sequencing, this method captures multiple layers of information—including chromatin accessibility, DNA methylation, protein footprints, and genetic variants — from ONE experiment. The result is a high-resolution view of chromatin regulation that cannot be achieved with traditional short-read approaches.
What is Fiber-seq?
Fiber-seq1 is a long-read sequencing assay that simultaneously maps genetic and epigenetic features from individual chromatin fibers. The method uses a methyltransferase (Hia5) to label accessible adenines by methylating them, creating N6-methyladenine (6mA), effectively “stenciling” chromatin accessibility onto DNA. Next, long-read technologies such as Pacific Biosciences® (PacBio®) HiFi or Oxford Nanopore Technologies® (ONT) sequencing detect both the stenciled 6mA and endogenous DNA methylation, producing multiomic data along individual DNA molecules. Projects that would normally need four different assays can now be completed in one Fiber-seq reaction (Table 1).

How does Fiber-seq stack against ATAC-seq, bisulfite sequencing, and ChIP-seq?
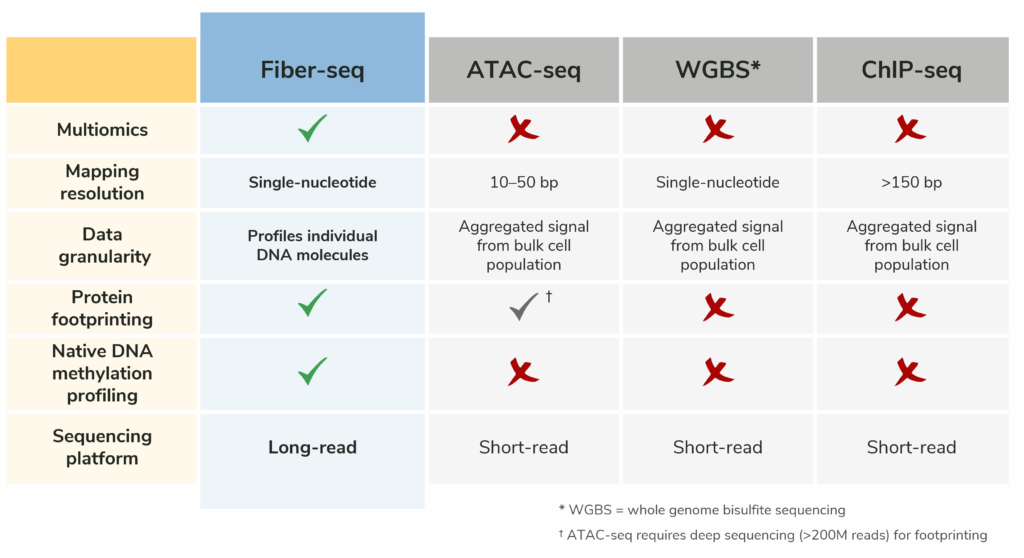
Table 2: Comparison of the key features of Fiber-seq with ATAC-seq, bisulfite sequencing, and ChIP-seq. Fiber-seq streamlines multiomic analysis by combining multiple assays into a single experiment and enables profiling of complex, hard-to-map genomic regions that are often missed by short read methods.
In a single experiment, Fiber-seq profiles (Figure 1):
- Chromatin accessibility
- DNA methylation (5mC, 5hmC)
- Protein footprints (including nucleosomes, transcription factors, and RNA polymerase)
- Genetic variants (SNPs, CNVs, and structural variants)
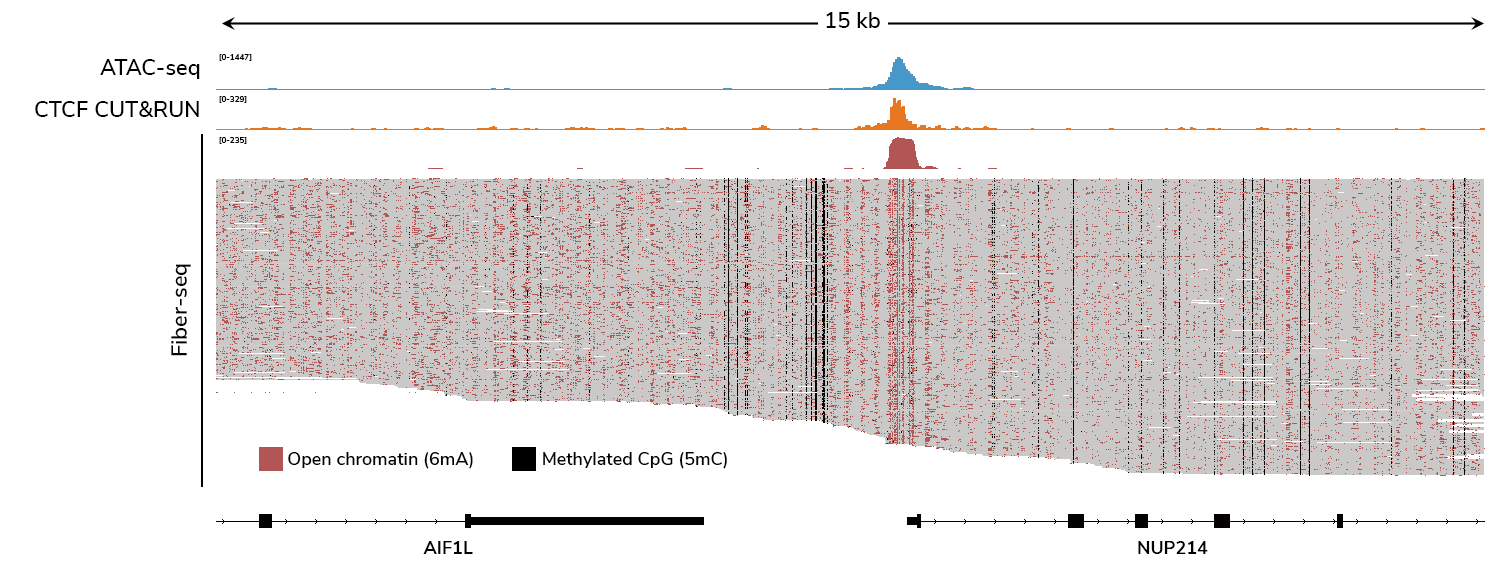
What does Fiber-seq data look like?
How does Fiber-seq work?
The Fiber-seq workflow is simple and scalable (Figure 2):
- Label open chromatin – Extract nuclei and briefly incubate them for 10 minutes with CUTANA™ Hia5, which methylates accessible adenines, creating 6mA.
- Prep library and sequence– Purify and prepare genomic DNA using standard long-read library prep protocols for either PacBio or Nanopore sequencing.
- Analyze long-read sequencing data – Long-read sequencing platforms detect both stenciled and endogenous DNA methylation, generating single-molecule maps of chromatin features. Researchers can also aggregate data for bulk analysis.
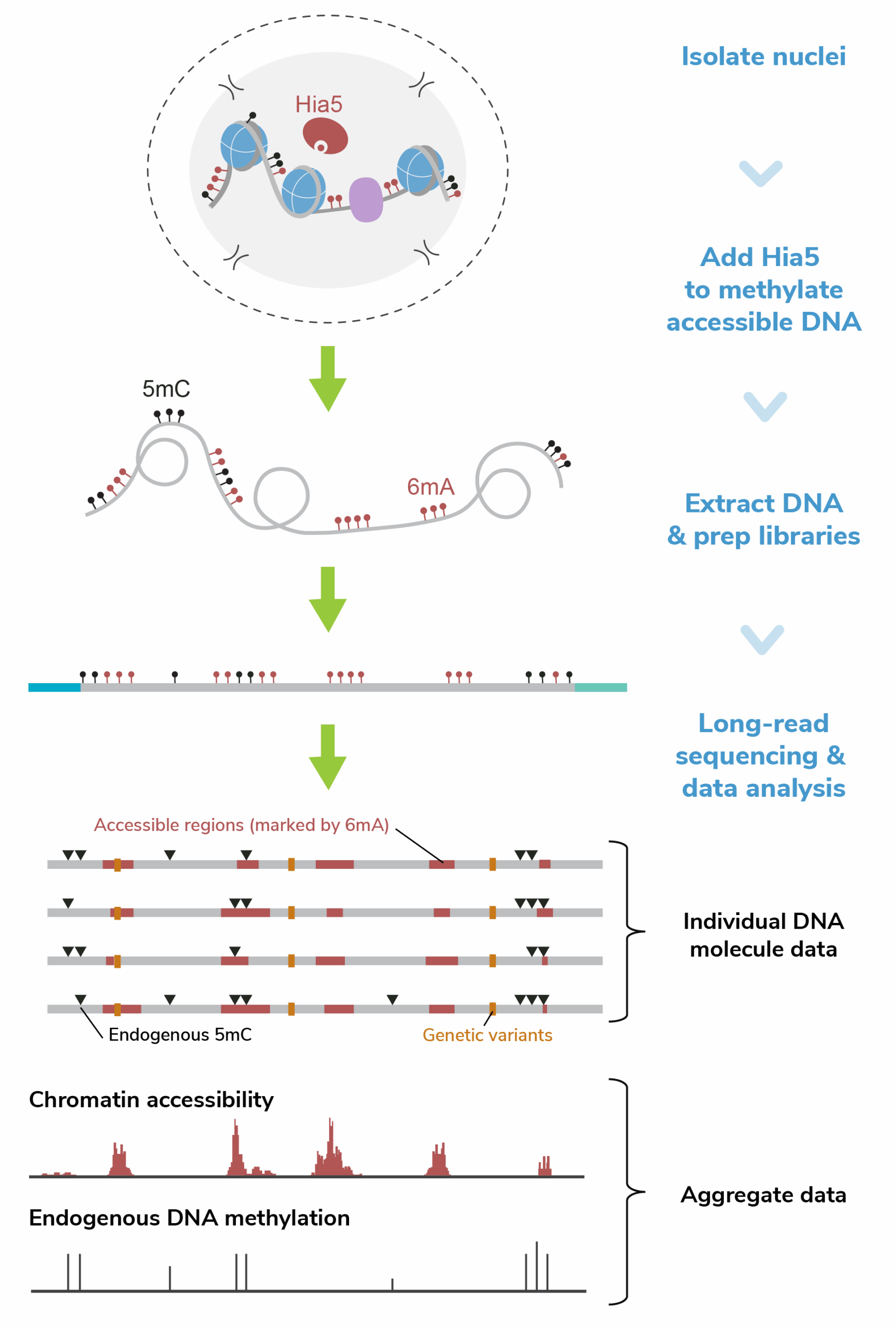
Because Fiber-seq does not require PCR amplification or antibodies, the workflow avoids common biases and reduces experimental complexity. Further, long-read sequencing enables better coverage of the genome (i.e., repetitive or structurally complex genomic regions) and allele-specific profiling of both genetic and epigenetic features (i.e., haplotype phasing).
Want to get started with Fiber-seq?
EpiCypher offers several CUTANA™ Fiber-seq products to get this assay up and running in your lab.
How is Fiber-seq better than ATAC-seq?
ATAC-seq works by using a hyperactive transposase (Tn5) to cleave accessible DNA and simultaneously insert adapters, followed by sequencing on short-read platforms. While this method is powerful for mapping open chromatin, it has several drawbacks. Detecting transcription factor or nucleosome footprints requires extremely deep sequencing2 (over 200 million reads for human genome or equivalent size). At such depths, it is hard to tell apart truly identical Tn5 insertion events from PCR duplicates created during library amplification3. As a result, real biological signals can be mistaken for artifacts, reducing the accuracy of footprinting and complicating analysis. Further, filtering out PCR duplicates can bias results towards highly accessible regions.
Fiber-seq overcomes these challenges by directly profiling individual DNA molecules. This provides much higher resolution for detecting protein footprints (Figure 3), including transcription factors, nucleosomes, and even RNA polymerase II4, all without the need for antibodies. Unlike ATAC-seq, Fiber-seq does not require averaging signals across a population of cells, revealing chromatin heterogeneity that bulk methods miss.
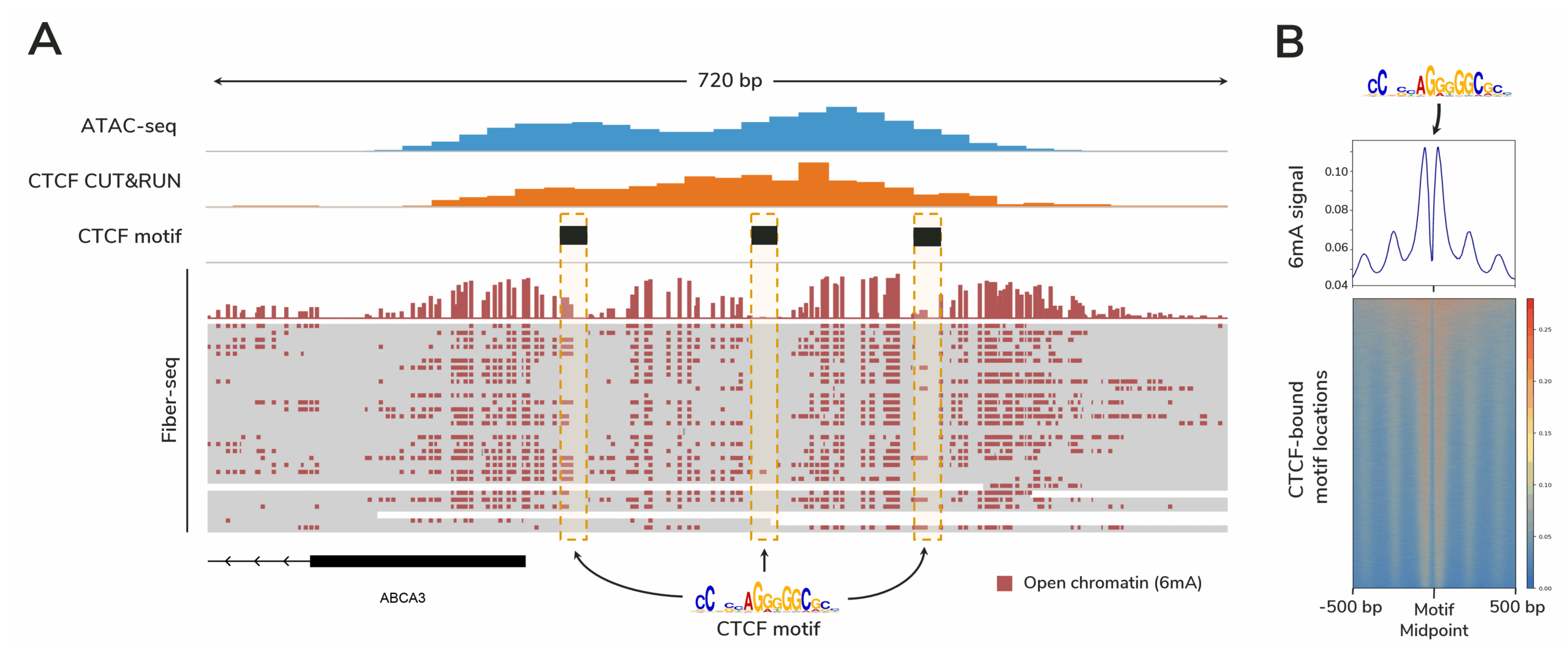
Figure 3. Fiber-seq profiles protein footprints with higher resolution than ATAC-seq.
Figure caption
(A) Fiber-seq data from 30 individual DNA molecules are shown, with the top track displaying aggregate 6mA signal marking open chromatin. Each horizontal line below this track represents a single DNA molecule. Dotted boxes indicate CTCF motif regions. Because Fiber-seq provides single-molecule resolution, heterogeneity in protein binding across the cell population is revealed (the leftmost boxed region shows variable 6mA labeling, suggesting differential CTCF occupancy).
(B) Genome-wide 6mA signal from this Fiber-seq experiment demonstrates robust footprinting of CTCF sites. Elevated 6mA levels flanking the motif midpoint reflect accessible DNA surrounding CTCF binding sites.
Since Fiber-seq is PCR-free, it avoids amplification biases and makes experiments both simpler and faster. In fact, the benchwork before sequencing can be completed in about half the time of an ATAC-seq experiment. And because it leverages long-read sequencing platforms, this approach also detects endogenous DNA methylation alongside accessibility and protein footprints, delivering multiple layers of information in one experiment.
Where is this assay most useful?
🌐 Multiomic research
Traditional multiomic studies often require performing separate assays such as ATAC-seq, ChIP-seq, and bisulfite sequencing. Fiber-seq profiles chromatin accessibility, protein footprints, DNA methylation, and genetic variants in a single experiment, providing high-resolution insights while reducing the need for multiple workflows.
⚕️ Clinical genetics and translational studies
Understanding how genetic variants influence chromatin regulation is critical for uncovering disease mechanisms. Because Fiber-seq uses long-read sequencing, relationships between variants and their associated epigenetic features are preserved across long stretches of DNA, enabling haplotype phasing. This makes Fiber-seq valuable for helping interpret variants of uncertain significance (VUSs) and for investigating Mendelian disorders.
💊 Drug discovery and development
Therapeutic response is often shaped by chromatin dynamics that standard methods cannot resolve. Fiber-seq accelerates drug development and clinical research by providing comprehensive views of multiple regulatory features in a single assay, without significantly increasing long-read sample processing time. Further, it reduces the number of separate assays required to understand a drug’s mechanism of action, identify off-target effects, or assess the functional impact of genetic variants.
🧩 Illuminating complex genomic regions
Repetitive regions such as telomeres, centromeres, and transposable elements remain difficult to study with short-read methods. Fiber-seq leverages long reads to resolve chromatin organization in these challenging regions, shedding light on genome stability and cell behavior. Such findings are especially important for fields ranging from cancer and aging to evolutionary biology.
🆕 Lowering the barrier to epigenomics research
For researchers new to chromatin biology, Fiber-seq offers a straightforward entry point. Its antibody-free, PCR-free workflow integrates seamlessly with long-read sequencing pipelines, eliminating the need for multiple specialized assays or complex protocols.
🦎 Emerging model organisms
In species without a high-quality reference genome, long-read sequencing enables accurate, contiguous assemblies. Fiber-seq can be directly used to generate de novo assemblies while also profiling DNA accessibility and methylation, adding layers of epigenetic information on top of new reference genomes. This opens the door to epigenomic studies in novel model species that were previously inaccessible.
What are applications of this tech?
Researchers are already using Fiber-seq to address diverse biological questions:
- Resolve rare disease – Link genetic variants to gene regulatory changes to uncover mechanisms of Mendelian disorders. PMID: 39880924.
- Profile brain epigenomics – Map cell-type-specific chromatin architecture in the human brain. PMID: 39631398.
- Unravel complex immunology – Identify haplotype-selective chromatin accessibility in immune cells. PMID: 40501892.
- Track transcriptional dynamics – Monitor RNA polymerase activity and its effects on chromatin structure. PMID: 39191261.
- Explore transposable elements – Reveal hidden regulatory elements in highly repetitive crop genomes PMID: 40360747.
- Illuminate centromeres – Characterize conserved chromatin organization in repetitive centromeric regions across species. PMID: 40147439.
References
- Stergachis, A. B., Debo, B. M., Haugen, E., Churchman, L. S. & Stamatoyannopoulos, J. A. Single-molecule regulatory architectures captured by chromatin fiber sequencing. Science 368, 1449-1454 (2020). https://doi.org/10.1126/science.aaz1646
- Yan, F., Powell, D. R., Curtis, D. J. & Wong, N. C. From reads to insight: a hitchhiker’s guide to ATAC-seq data analysis. Genome Biol 21, 22 (2020). https://doi.org/10.1186/s13059-020-1929-3
- Zhu, T., Liao, K., Zhou, R., Xia, C. & Xie, W. ATAC-seq with unique molecular identifiers improves quantification and footprinting. Commun Biol 3, 675 (2020). https://doi.org/10.1038/s42003-020-01403-4
- Tullius, T. W. et al. RNA polymerases reshape chromatin architecture and couple transcription on individual fibers. Mol Cell 84, 3209-3222 e3205 (2024). https://doi.org/10.1016/j.molcel.2024.08.013
Spin up your research with CUTANA™ Fiber-seq Today!
Ready to Unravel the Epigenome with Long-Read Sequencing? EpiCypher offers solutions to bring this approach into your research:
- CUTANA™ Fiber-seq Kit: Everything you need to go from cells to library prep-ready DNA. Every kit comes with a manual packed with detailed protocols, QC recommendations, FAQs, and expert tips — plus a bench-proof QuickStart card and access to the Tech Support Center for additional guidance
- CUTANA™ Hia5 Enzyme: the key enzyme required for Fiber-seq. Ideal for DIY users or those running custom workflows.
- CUTANA™ Fiber-seq Services: Prefer to have our experts run experiments for you? Explore our services for professional assay support and data delivery.