Genome-Wide DNA Methylation Mapping
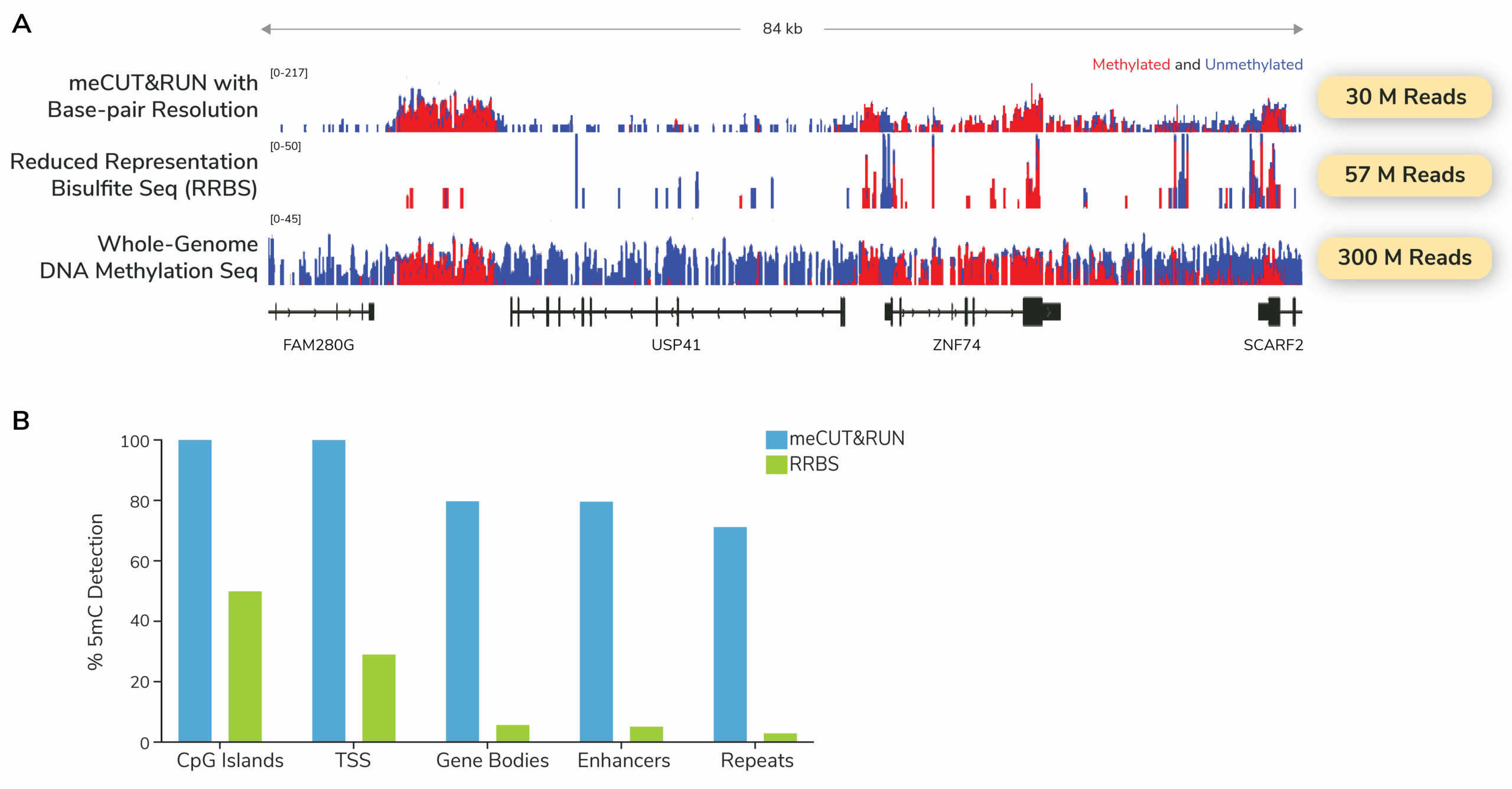
Powered by meCUT&RUN, this approach enables scalable whole-genome DNA methylation profiling while requiring ~20-fold less sequencing than whole-genome bisulfite sequencing. This makes meCUT&RUN ideally suited for discovery and perturbation studies.
Targeted DNA Methylation Profiling
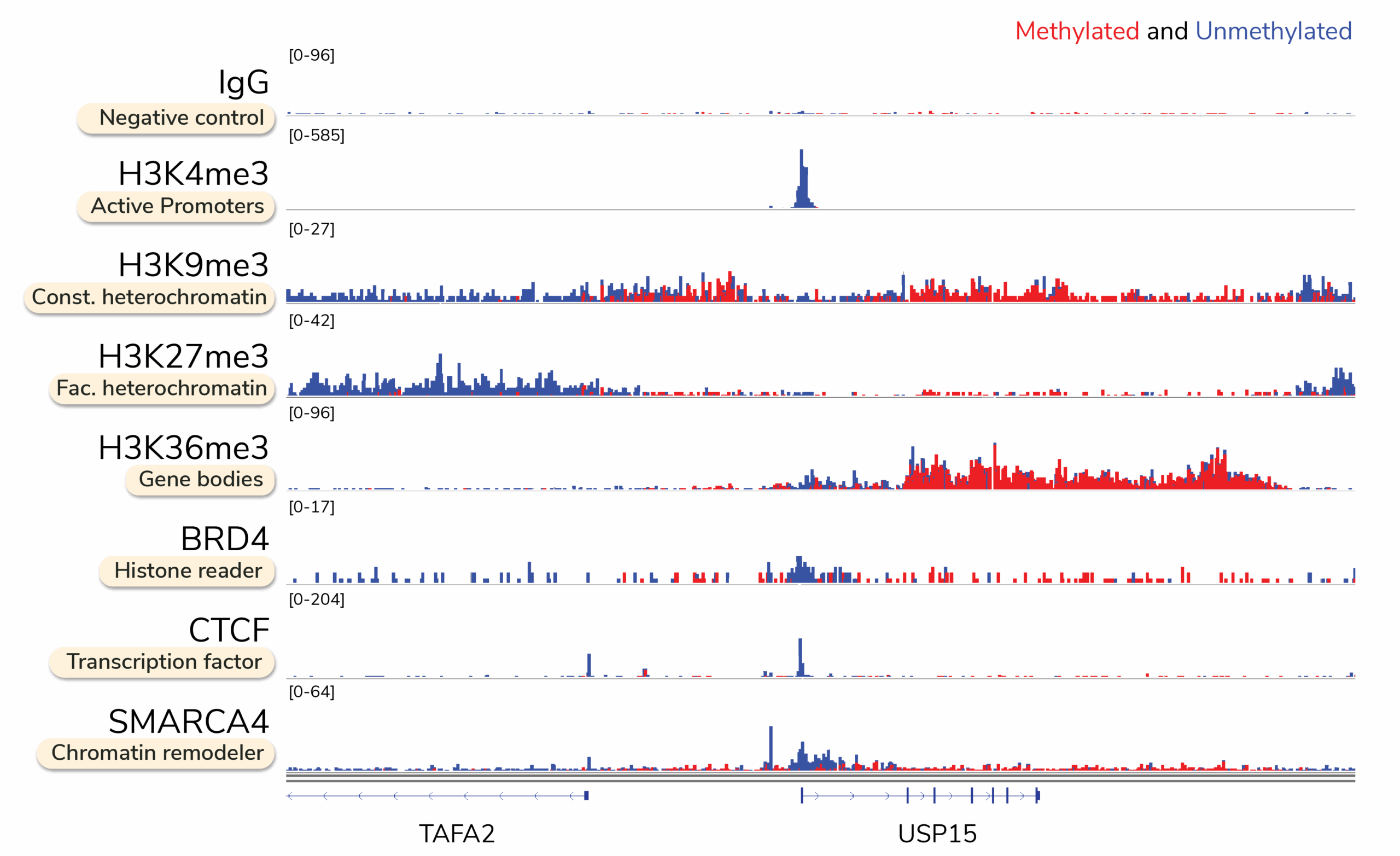
Multiomic CUT&RUN profiles DNA methylation directly at targeted chromatin proteins, enabling causal linkage between methylation changes and regulatory element activity. This integrated approach is well suited for mechanism-of-action studies, where understanding how DNA methylation influences chromatin regulation is essential.
CUTANA™ Fiber-seq: long-read multiomics
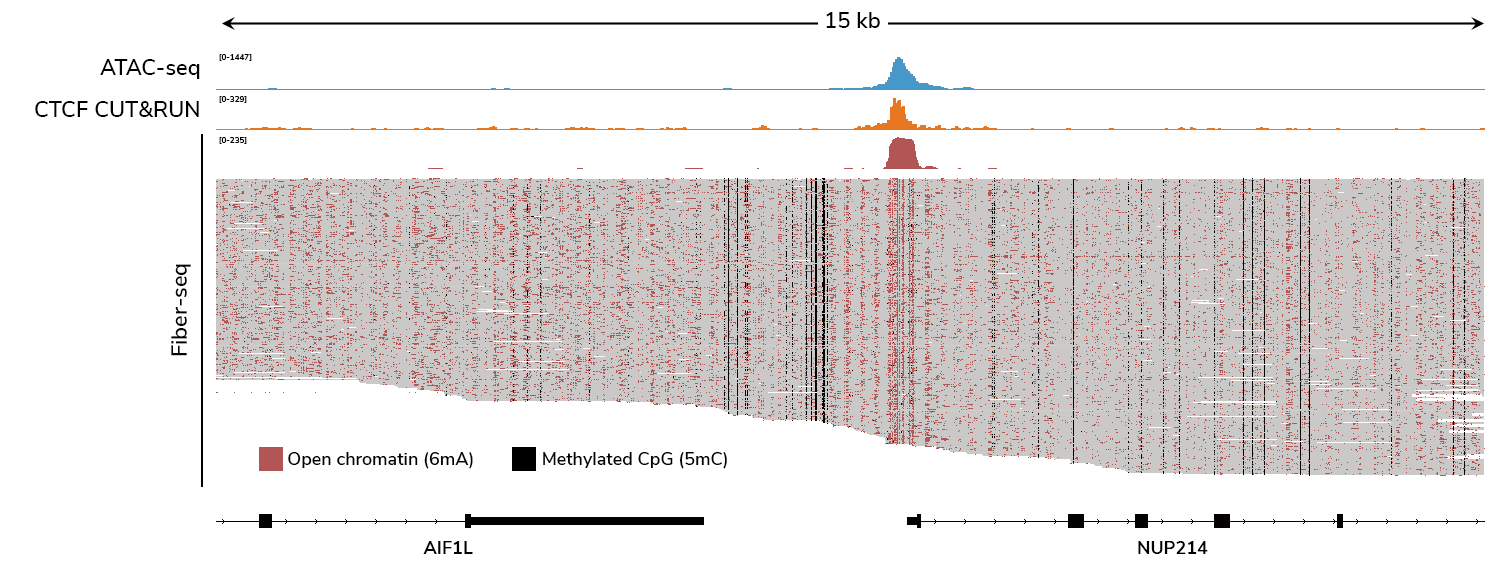
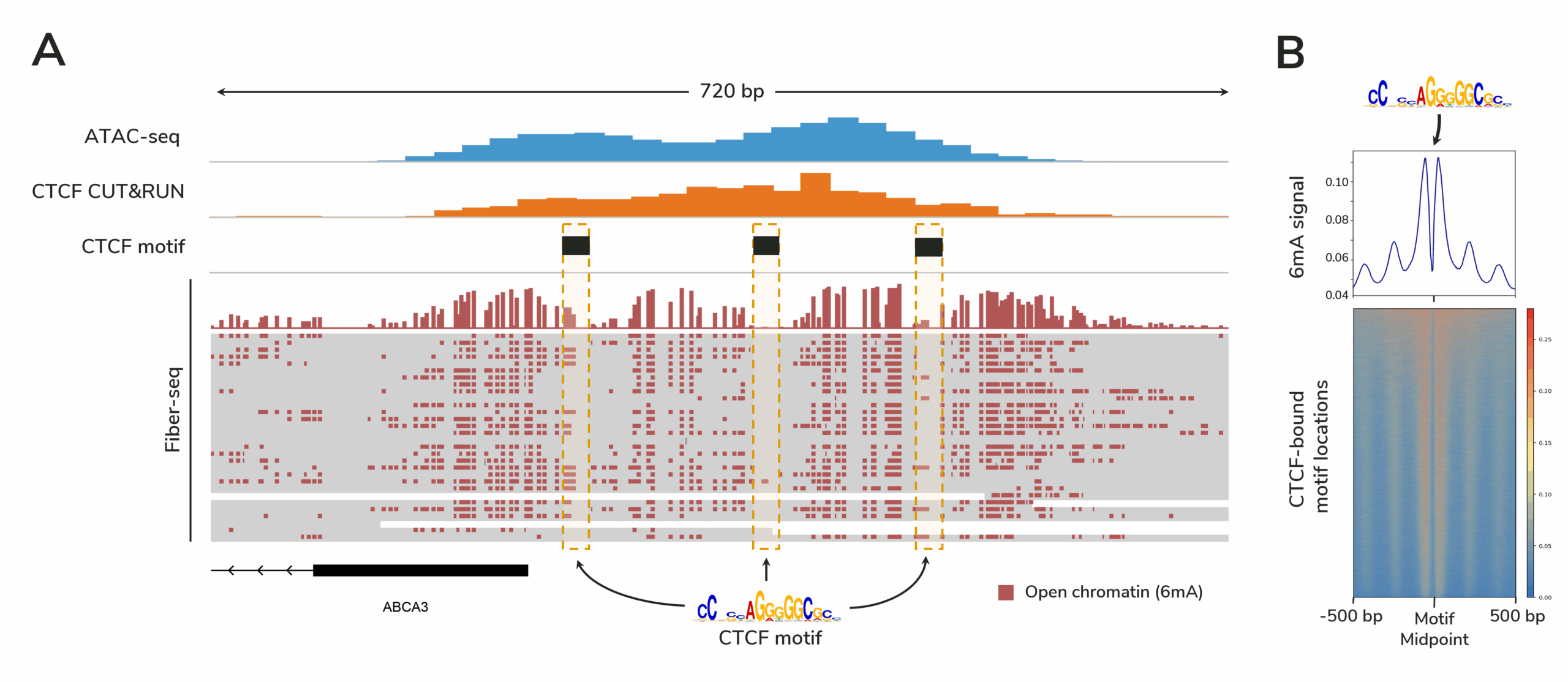
Fiber-seq provides a comprehensive view of gene regulation by simultaneously profiling chromatin accessibility, DNA methylation, protein footprints, and genetic variation in a single long-read sequencing assay. By delivering single-molecule resolution, Fiber-seq reveals epigenomic insights in complex and heterogeneous samples that bulk assays cannot resolve.
What are the differences between the DNA methylation assay options?
How do I decide which DNA methylation assay to choose?
What does whole-genome methylation sequencing data look like with meCUT&RUN?
What does targeted DNA methylation mapping data look like with Multiomic CUT&RUN?
What does Fiber-seq data look like? What other insights does it provide?