A Comprehensive Guide to DNA Methylation Sequencing Methods
- Ellen Weinzapfel
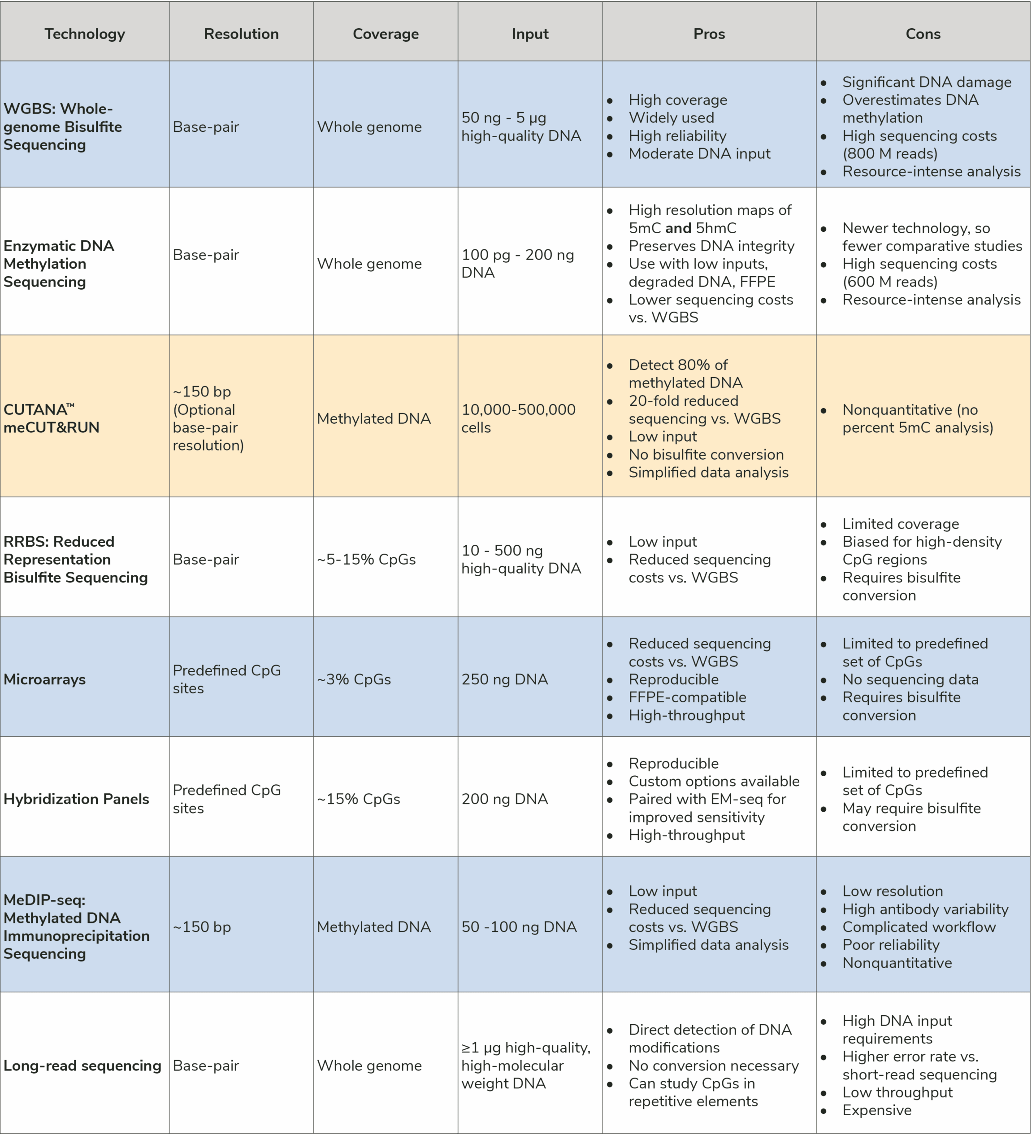
DNA methylation is a critical epigenetic modification with profound implications on gene regulation, development, and disease research. As interest in epigenomics grows, researchers require robust and reliable methods for DNA methylation sequencing. However, choosing the right technology can be challenging due to differences in resolution, coverage, sample requirements, and cost. This guide explores the leading DNA methylation sequencing methods, their strengths and limitations, and the ideal applications for each (Table 1). Key publications highlighting their development and use are also included.
1. Whole Genome Bisulfite Sequencing (BS-seq, WGBS)
Overview: Bisulfite sequencing is the gold standard for genome-wide DNA methylation analysis. In this strategy, sodium bisulfite is used to convert unmethylated cytosines to uracil, while methylated and hydroxymethylated cytosines remain unchanged. The result is base-pair resolution DNA methylation sequencing across the entire genome.
Pros: Base-pair resolution; high coverage; widely used.
Cons: Harsh chemical treatment degrades DNA; requires deep sequencing for comprehensive results; potential overestimation of DNA methylation; resource-intensive computational processing.
Best For: Whole-genome methylation analysis in high-quality DNA samples.
Relevant Articles:
Original bisulfite conversion article:
- Frommer et al. PNAS 1992. doi.org/10.1073/pnas.89.5.1827
First genome-wide, base-pair resolution bisulfite sequencing (Arabidopsis thaliana):
- Cokus et al. Nature 2008. doi.org/10.1038/nature06745
- Lister et al. Cell 2008. doi.org/10.1016/j.cell.2008.03.029
First genome-wide bisulfite sequencing of the human genome:
- Lister et al. Nature 2009. doi.org/10.1038/nature08514
Deep whole-genome bisulfite sequencing of multiple human cell types:
- Loyfer et al. Nature 2023. doi.org/10.1038/s41586-022-05580-6
2. Enzymatic DNA Methylation Profiling Strategies
Overview: Enzymatic DNA methylation profiling uses a series of enzymatic reactions, instead of bisulfite conversion, to selectively convert unmethylated cytosines to uracil. These alternative whole-genome DNA methylation sequencing strategies are gentler than bisulfite conversion, which helps preserve DNA integrity, and reduces sequencing requirements. Additional enzymes can be used to further distinguish between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC), an exciting innovation in the DNA methylation profiling toolbox.
Pros: Base-pair resolution of 5mC and 5hmC; vs. bisulfite conversion has reduced DNA damage, better performance with low-input samples; potentially compatible with formalin-fixed paraffin-embedded (FFPE) tissues.
Cons: Still requires fairly deep sequencing; resource-intensive computational processing; relatively new, so fewer comparative studies.
Best For: High-precision DNA methylation profiling in low-input or degraded samples.
Relevant Articles:
Development and comparison with whole-genome bisulfite sequencing:
- Vaisvilla et al. Genome Research 2021. doi.org/10.1101/gr.266551.120
- Feng et al. Epigenetics & Chromatin 2020. doi.org/10.1186/s13072-020-00361-9
- Morrison et al. Epigenetics & Chromatin 2021. doi.org/10.1186/s13072-021-00401-y
- Guerin et al. Cell Reports 2025. doi.org/10.1016/j.celrep.2025.115680
Overview: meCUT&RUN is an innovative DNA methylation profiling assay based on ultra-sensitive CUT&RUN (Cleavage Under Targets & Release Using Nuclease) and EpiCypher’s groundbreaking Reader CUT&RUN technology. meCUT&RUN uses a methyl-binding domain to selectively enrich methylated DNA, followed by traditional library prep or cytosine conversion library prep (for base-pair resolution). meCUT&RUN captures 80% of DNA methylation using just 20-50 M reads, offering a low-cost, high-resolution option for 5mC analysis.
Pros: 20-fold less sequencing vs. WGBS makes assay highly affordable; capture 80% of DNA methylation, including CpG sites in enhancers, promoters, gene bodies, and repetitive elements; robust down to 10,000 cells; optional base-pair resolution.
Cons: Nonquantitative, no percent DNA methylation analysis.
Best For: Cost-sensitive whole-genome studies where base-pair resolution isn’t required; identifying DNA methylation patterns at key regulatory regions.
Relevant Articles:
Development of Reader CUT&RUN assays:
- Marunde et al. eLife 2024. doi.org/10.7554/eLife.78866
4. Reduced Representation Bisulfite Sequencing (RRBS)
Overview: RRBS is a low-cost, base-pair resolution DNA methylation profiling strategy that was developed as an alternative to microarrays and enrichment-based methods. RRBS uses methylation-specific restriction enzyme (MSRE) digestion to generate genomic fragments, followed by bisulfite conversion for base-pair resolution sequencing data.
Pros: Cost-effective; focused analysis of CpG islands and gene promoters, which are often hypermethylated in cancer and represent important potential biomarkers.
Cons: Limited genome coverage (~5-10% of CpGs); biased for areas with high CpG density.
Best For: Cost-sensitive studies focusing on CpG islands and promoters.
Relevant Articles:
Original RRBS article:
- Meissner et al. Nucleic Acids Research 2005. doi.org/10.1093/nar/gki901
Application in stem cells vs. differentiated cells:
- Meissner et al. Nature 2008. doi.org/10.1038/nature07107
Updated single-cell RRBS protocol:
- Guo et al. Nature Protocols 2015. doi.org/10.1038/nprot.2015.039
5. DNA Methylation Profiling Microarrays
Overview: DNA methylation arrays integrate bisulfite conversion with hybridization to sets of oligonucleotide probes, allowing detection of DNA methylation levels at specific CpG sites. Newer arrays by Illumina cover >900,000 CpG sites and are designed for high-throughput processing. These microarrays help offset the high costs associated with next-generation sequencing and downstream data analysis, making them an accessible and cost-effective solution for clinical research and labs without bioinformatics expertise.
Pros: Cost-effective for large sample sets; well-established, high-throughput technology; high reproducibility; compatible with FFPE tissues.
Cons: Limited to predefined CpG sites; favor CpG islands; no sequencing data.
Best For: Large-scale studies where predefined CpG site coverage is sufficient, such as epidemiological research or biomarker discovery.
Relevant Articles:
Early DNA methylation array development:
- Ishkanian et al. Nature Genetics 2004. doi.org/10.1038/ng1307
- Bibikova et al. Genome Research 2006. doi.org/10.1101/gr.4410706
- Bibikova et al. Epigenomics 2009. doi.org/10.2217/epi.09.14
- Sandoval et al. Epigenetics 2011. doi.org/10.4161/epi.6.6.16196
Key studies demonstrating application of DNA methylation profiling arrays for cancer research:
- Capper et al. Nature 2018. doi.org/10.1038/nature26000
- Bailey et al. Cancer Cell 2018. doi.org/10.1016/j.cell.2018.02.060
6. Methylated DNA Immunoprecipitation Sequencing (MeDIP-seq)
Overview: MeDIP-seq uses a 5-methylcytosine antibody to selectively enrich methylated DNA fragments from a pool of sheared DNA, followed by next-generation sequencing for genome-wide analysis. Isolation of methylated DNA allows researchers to study whole-genome DNA methylation patterns with reduced sequencing depths compared to bisulfite sequencing or enzymatic conversion strategies. However, using immunoprecipitation combined with poor quality 5-methylcytosine antibodies creates multiple problems with reliability, resolution, and accuracy.
Pros: Cost-effective at only ~30 million reads; useful for genome-wide methylation analysis; easier to analyze compared to bisulfite or enzymatic conversion methods.
Cons: Low resolution and high background; high variability; poor antibody quality; biased toward highly methylated regions.
Best For: Studying genome-wide methylation trends rather than single-site resolution.
Relevant Articles:
Early application of MeDIP combined with microarray analysis to study DNA methylation across human genome:
- Weber et al. Nature Genetics 2005. doi.org/10.1038/ng1598
Ultra-low input protocol development:
- Taiwo et al. Nature Protocols 2012. doi.org/10.1038/nprot.2012.012
Comparison with other DNA methylation profiling techniques:
- Down et al. Nature Biotechnology 2008. doi.org/10.1038/nbt1414
- Harris et al. Nature Biotechnology 2010. doi.org/10.1038/nbt.1682
- Beck, Ben Maamar & Skinner. Epigenetics 2022. doi.org/10.1080/15592294.2021.1924970
7. Long-Read Sequencing (PacBio/Nanopore)
Overview: Long-read sequencing technologies, such as PacBio and Oxford Nanopore, provide direct detection of DNA methylation on native DNA, without requiring bisulfite or enzymatic conversion. The ability to detect DNA methylation on long kilobase-length (or longer) fragments allows researchers to map DNA methylation in repetitive regions that are inaccessible by short-read sequencing, revealing the interplay between 5-methylcytosine and structural variants.
Pros: Direct methylation detection; long-read capability enables phasing of DNA modifications with haplotypes; minimal sample processing.
Cons: Historically, long-read platforms require more DNA, are lower throughput, and show higher error rates vs. short-read sequencing; fewer established data analysis pipelines.
Best For: Phasing DNA methylation with genetic variants; studying methylation patterns in repetitive regions and structural variants.
Relevant Articles:
PacBio-based papers and applications:
- Flusberg et al. Nature Methods 2010. doi.org/10.1038/nmeth.1459
- Wenger et al. Nature Biotechnology 2019. doi.org/10.1038/s41587-019-0217-9
- Tse et al. PNAS 2021. doi.org/10.1073/pnas.2019768118
- Cheung et al. Nature Communications 2023. doi.org/10.1038/s41467-023-38782-1
- Ni et al. Nature Communications 2023. doi.org/10.1038/s41467-023-39784-9
Oxford Nanopore-based papers and applications:
- Simpson et al. Nature Protocols 2017. doi.org/10.1038/nmeth.4184
- Rand et al. Nature Communications 2017. doi.org/10.1038/nmeth.4189
- Liu et al. Nature Communications 2019. doi.org/10.1038/s41467-019-10168-2
- Stefansson et al. Nature Genetics 2024. doi.org/10.1038/s41588-024-01851-2
Choosing the Right Technology: Which DNA Methylation Sequencing Assay Fits Your Needs?
Selecting the best DNA methylation profiling method depends on several factors (Table 1), including:
- Resolution needs: Is base-pair resolution necessary, or is regional DNA methylation data sufficient?
- Coverage requirements: Whole-genome vs. site-specific analysis.
- Sample quality: Some methods are better for degraded or low-input samples.
- Number of samples: Large-scale studies may benefit from microarrays or targeted sequencing approaches.
- Budget: Whole-genome sequencing methods are more costly than targeted approaches.
- Research goals:
- Functional genomics: Genome-wide approaches help identify novel mechanisms.
- Biomarker discovery: Targeted methods can balance coverage with high throughput capabilities.
- Disease profiling: Method selection varies based on sample type and/or quality.
Conclusion
Advancements in DNA methylation profiling are expanding research possibilities, enabling more precise and high-throughput analysis. While bisulfite sequencing remains a gold standard, emerging technologies like EM-seq and long-read sequencing offer promising alternatives for different applications. By understanding the strengths and limitations of each method, researchers can select the best approach to advance their studies.
Are you interested in learning more about our DNA methylation assays? Request a quote below!