The Complete Guide to CUT&Tag Experiments
Table of Contents
- What is CUT&Tag?
- How is CUT&Tag different from ChIP-seq?
- How does CUT&Tag compare to CUT&RUN? Which is best for my project?
- The 8 Basic Steps of CUT&Tag
- How to Design and Optimize CUT&Tag Assays
- Next Steps for Getting Started with CUT&Tag
What is CUT&Tag?
CUT&Tag, or Cleavage Under Targets and Tagmentation, is an innovative epigenomic mapping strategy that builds on its predecessor immunotethering technologies CUT&RUN and ChIC1-6. The CUT&Tag assay was developed by Dr. Steven Henikoff’s lab and first reported in 20195. In CUT&Tag, a fusion of Protein A, Protein G, and transposase Tn5 (pAG-Tn5) is used to selectively cleave and ligate sequencing adapters at antibody-bound chromatin loci in intact nuclei (or cells). Target chromatin fragments are amplified using primers that recognize the adapter-ligated DNA, purified, and sequenced, generating high quality data with low background.
Since its initial publication, CUT&Tag has emerged as a powerful assay for mapping histone post-translational modifications (PTMs) and select transcription factors from small numbers of cells, including single cells5-13. CUT&Tag often draws comparisons to ChIP-seq and CUT&RUN, as all three assays apply next-generation sequencing (NGS) to profile genome-wide enrichment of selected chromatin targets. The first sections of this blog will draw proper distinctions between the three assays and ideal applications of CUT&Tag compared to CUT&RUN. Then we will review the basic steps of CUT&Tag, experimental design, and key optimization steps.
Start your assay now with CUTANA™ CUT&Tag Reagents
How is CUT&Tag different from ChIP-seq?
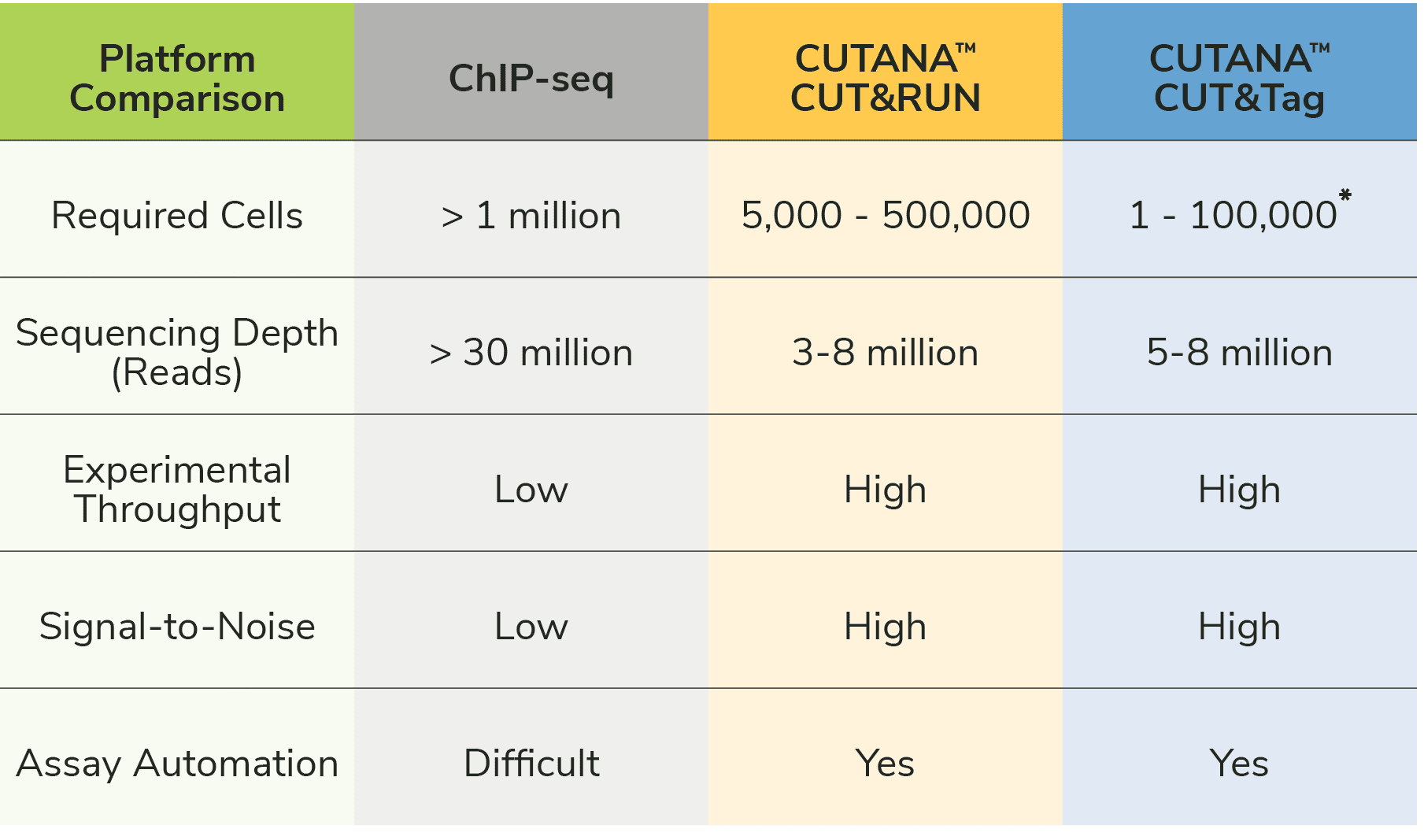
CUT&Tag bypasses the most challenging and time-consuming parts of ChIP-seq assays, including cross-linking, chromatin fragmentation, and immunoprecipitation (IP). Eliminating these steps provides multiple advantages (Figure 1):
- Faster: The streamlined workflow can go from cells to sequence-ready libraries in two days, compared to five days (or more) with ChIP-seq.
- Fewer cells. CUTANA™ CUT&Tag assays generate high-resolution profiles using as few as 10,000 nuclei and single cell applications have been reported multiple times in the literature5-13. ChIP-seq requires millions of cells for high quality sequencing, which has hindered analysis of single cells and/or precious samples.
- Improved signal-to-noise. Separation of target-bound chromatin from intact nuclei results in lower background and robust signal – even when using fewer starting cells.
- No library prep. By adding sequencing adapters at antibody-bound target sites, CUT&Tag can skip traditional library prep steps (end repair, adapter ligation), which greatly simplifies the protocol. EpiCypher’s CUTANA CUT&Tag Protocol further streamlines this strategy by amplifying tagmented DNA directly from the reaction mixture, allowing users to go from cells to PCR amplified libraries in one tube6,7.
- Reduced cost. CUT&Tag assays only require 5-8 million sequencing reads for confident peak calling, driving >70% cost savings compared to ChIP-seq (requires ~30 million reads).
- Higher throughput. The entire assay is performed using intact nuclei immobilized on magnetic beads, enabling high throughput and adaptation for automated systems14.
Note: Automation of ChIP-seq is impractical due to its high cost, optimization requirements, low throughput, and poor data quality, meaning ChIP-seq has minimal applications in clinical research and drug development. Automated CUT&Tag provides a major breakthrough approach for this area of research!
How does CUT&Tag compare to CUT&RUN? Which is best for my project?
We discussed CUT&RUN in a previous blog. Both CUT&RUN and CUT&Tag use pAG to "tether" enzymes at antibody-labelled chromatin in intact cells/nuclei, which enables high quality profiling using low cells numbers and reduced sequencing costs (vs. ChIP-seq; Figure 1). The main difference between the two assays is the enzyme fused to pAG: CUT&RUN applies micrococcal nuclease (pAG-MNase) to cleave antibody-bound chromatin, while CUT&Tag uses pAG-Tn5 to simultaneously fragment and ligate sequencing adapters.
CUT&RUN requires traditional library prep steps, making the protocol approximately one day longer than CUT&Tag. The extra steps may increase the risk of sample loss, leading to slightly higher cell requirements in CUT&RUN (500,000 cells) vs. CUT&Tag (100,000 nuclei). When profiling from ultra-low cell numbers and/or if assay speed is important, CUT&Tag may be the ideal choice.
It is also important to consider assay compatibility with the experimental target (histone PTM or protein). CUTANA™ CUT&Tag is reliable for mapping histone PTMs, but is NOT recommended for chromatin-associated proteins (e.g. transcription factors). For chromatin-bound proteins we recommend our CUTANA™ CUT&RUN assays, which generate robust profiles for diverse protein and histone PTM targets. We also suggest CUT&RUN assays for scientists new to chromatin mapping because it is very reliable, requires few optimization steps, and is supported by our CUT&RUN Kit and Library Prep Kits.
For additional information, be sure to review our blog post on selecting the best assay for your experiment, and this post on assay optimization.
Go to the CUTANA™ CUT&Tag Protocol
The 8 Basic Steps of CUT&Tag
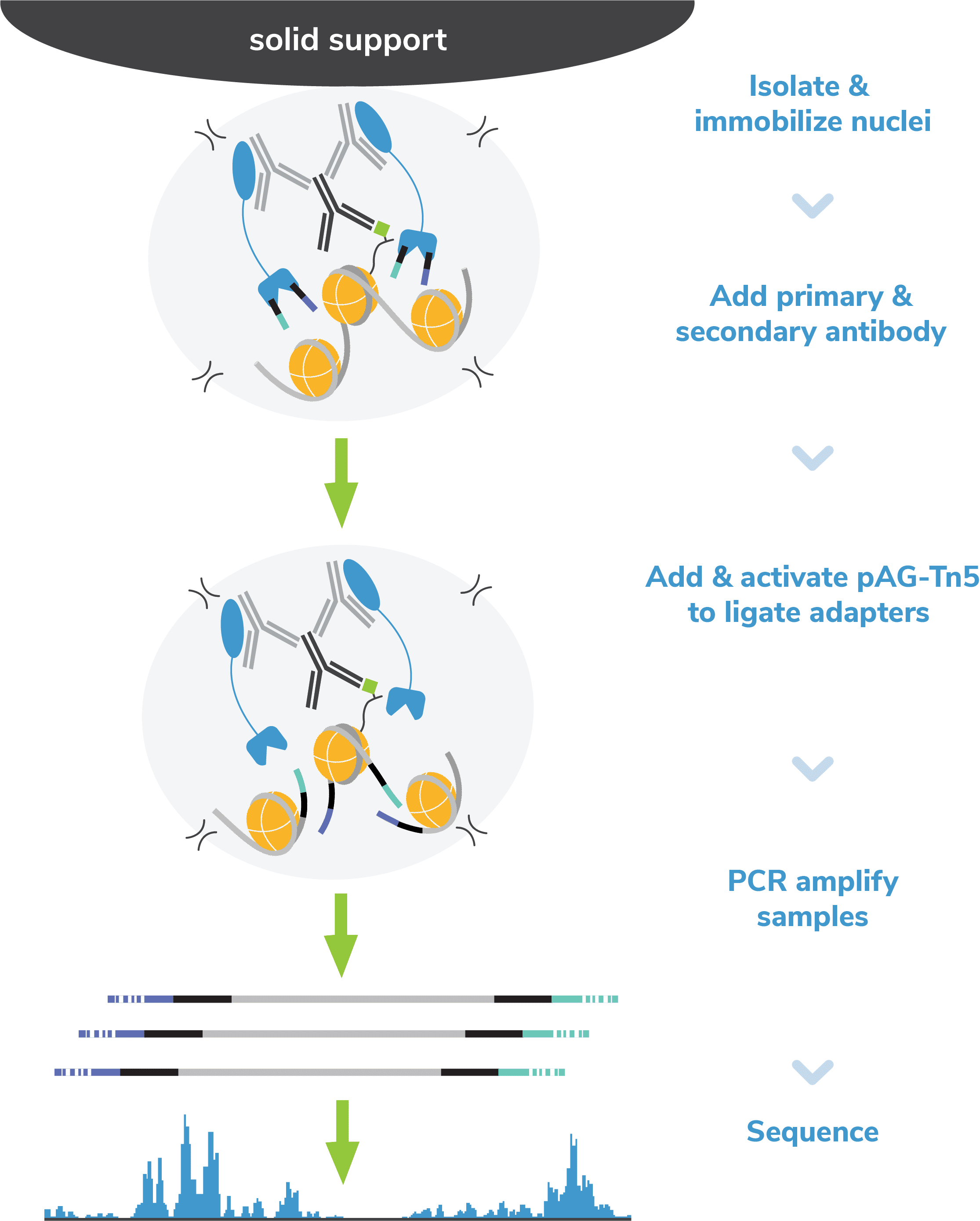
Step 1: Isolate nuclei and immobilize to magnetic beads.
Nuclei are the preferred input for CUT&Tag assays (see
Sample preparation). Nuclei are prepared from bulk cell populations and coupled to magnetic
beads coated with Concanavalin A (ConA; Figure 2). ConA is a
lectin that binds glycoproteins, glycolipids, and sugars found in the nuclear
membrane. Immobilization of nuclei on magnetic beads allows the use of 8-strip
tubes and multi-channel pipettes, which supports rapid sample processing and
improves assay reliability.
Important notes: It is critical to avoid ConA bead dry-out, which results in sample loss and reduced yields.
Step 2: Incubate with target-specific primary antibody.
An antibody specific to the histone PTM of interest is added to the reaction
and incubated overnight at 4˚C. EpiCypher always includes reactions with a
negative control (IgG,
EpiCypher 13-0042) and positive control (e.g. H3K27me3,
EpiCypher 13-0055) antibody in every experiment.
CRITICAL STEP: Selection of a highly specific primary antibody is crucial to CUT&Tag success, as off-target binding can significantly impact assay yields and data quality15,16. See How to Design and Optimize CUT&Tag Assays.
Step 3: Incubate with a species-specific secondary antibody.
The following day, reactions are washed and labelled with a species-matched
secondary antibody (e.g. anti-rabbit, anti-mouse), which amplifies pAG-Tn5
localization and subsequent on-target signal in sequencing data. At the end of
this step, reactions are washed with a high-salt buffer to remove unbound
antibodies and prepare for tagmentation.
CRITICAL STEP: The high-salt wash is essential to minimize nonspecific binding of pAG-Tn5 to accessible DNA, which results in ATAC-seq like signal in CUT&Tag datasets7.
Important notes: The cell-bead mixture often becomes clumpy after overnight incubation at 4'C, but can be dispersed by pipetting. Use low-retention filter tips and pipette gently to avoid nuclear lysis, which can increase bead clumping.
Step 4: Incubate with pAG-Tn5.
pAG-Tn5, pre-loaded with sequencing adapters, is added to each reaction and
binds antibody-labeled chromatin via the immunoglobulin binding properties of
pAG (Figure 2). Following incubation, the reactions are
washed several times using a high-salt buffer to remove nonspecific pAG-Tn5.
Step 5: pAG-Tn5 activation and targeted tagmentation
Tn5 is activated by addition of magnesium to cleave and ligate sequencing
adapters proximal to antibody-bound DNA (i.e. tagmentation). To help prevent
nonspecific Tn5 cleavage in open chromatin, tagmentation is performed under
high salt concentrations.
Tagmentation is quenched using a buffer containing EDTA, and DNA fragments are released into solution by heated digestion in a SDS buffer. SDS is subsequently neutralized using a nonionic detergent (i.e. Triton-X 100).
CRITICAL STEP: SDS inhibits indexing PCR – be sure to quench SDS activity!
Step 6: Indexing PCR
In EpiCypher’s exclusive
Direct-to-PCR CUT&Tag Protocol, indexing PCR is performed directly on digested chromatin from
Step 5 to generate NGS libraries (see note). The indexing
primers anneal to ligated adapter sequences, ensuring selective amplification
of tagmented DNA – even in the presence of cell debris. PCR parameters are
specifically optimized for mononucleosome-sized fragments, and primers contain
barcodes (or indexes) to enable multiplexed sequencing. This strategy allows
you to go from cells to PCR amplified sequencing libraries in a single tube,
minimizing sample loss and streamlining library prep.
Important note: The ConA beads are not removed and there is no DNA purification step prior to PCR. The number of PCR cycles may require optimization; see How to Design and Optimize CUT&Tag Assays.
Step 7: Library cleanup and quality analysis
CUT&Tag sequencing libraries are purified using SPRI magnetic beads. Library
concentration is determined using a fluorometric assay (e.g. ThermoFisher
Qubit™) and fragment size distribution is examined by capillary
electrophoresis (e.g. Agilent Bioanalyzer® or
TapeStation®). The PCR parameters in this protocol amplify
fragments compatible with Illumina® paired-end sequencing, with an
average fragment size of ~300 bp (~170 bp nucleosome + sequencing adapters;
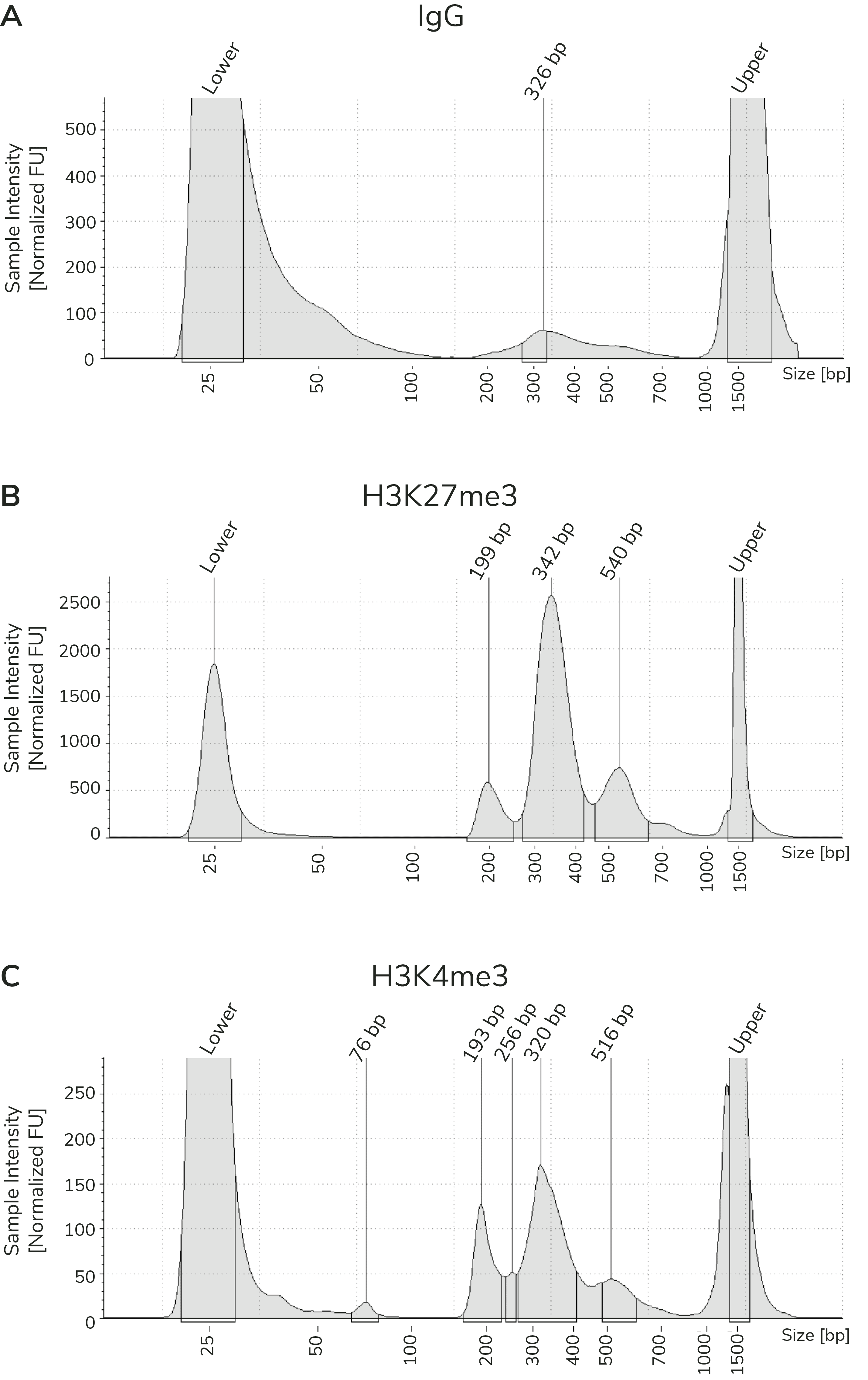
Figure 3). When comparing library yields and distribution
from CUTANA™ CUT&Tag assays, keep the following points in mind:
- The BEST indicator of CUT&Tag experimental success prior to sequencing is predominant enrichment of mononucleosome-sized fragments (~300 bp) in Bioanalyzer or TapeStation traces (Figure 3).
- Library yields should NOT be used as a definitive metric of success because results vary by cell type, target abundance, antibody specificity, number of nuclei, and so forth. Instead, EpiCypher recommends aiming for ~30 ng total DNA, which allows accurate library quantification, minimizes PCR duplicates, and enables library pooling at standard concentrations.
Step 8: Next-generation sequencing (NGS)
Sequencing libraries are pooled at equimolar ratios and sequenced on an
appropriate Illumina® sequencing platform. CUT&Tag only requires
5-8 million paired-end reads to generate high quality genomic maps, compared
to more than 30 million with ChIP-seq. As a result, users can multiplex more
libraries per sequencing run, enabling higher throughput and lower costs.
How to Design and Optimize CUT&Tag Assays
By selectively targeting antibody-bound chromatin in intact nuclei, CUT&Tag can generate high-resolution profiles with low background, exquisite reliability, and rapid throughput. However, even with EpiCypher’s robust Direct-to-PCR CUT&Tag Protocol, there are key optimization steps and control strategies to keep in mind. We have broken these guidelines into four main steps: Sample preparation, assay controls, antibody & target selection, and indexing PCR & sequencing.
I. Sample preparation
High quality sample prep is essential to CUT&Tag workflows.
Sample type: The preferred input for CUT&Tag is freshly isolated, unfixed (i.e. native) nuclei. Using nuclei in CUT&Tag reduces background signal from mitochondrial DNA in the cytoplasm and enables low sequencing depths (5-8 million reads). In addition, nuclei do not require optimization of permeabilization conditions or other alterations for unique cell types (e.g. immune cells, which can be activated by ConA). The CUTANA™ CUT&Tag Protocol starts with a nuclear isolation step and includes several quality control checks to evaluate sample quality (see Figure 4).
Notes on cell types and frozen materials: EpiCypher has developed modifications to our standard CUT&Tag protocol for whole cells, adherent cells, tissues, immune cells, and frozen nuclei/cells. Note that tissues must be processed into a mono-dispersion of cells. See the FAQs section of the CUTANA™ CUT&Tag Protocol for more information.
Considerations for using whole cells: If using cells, it is critical to optimize the digitonin concentration in CUT&Tag buffers for efficient permeabilization. See the CUTANA™ CUT&RUN Kit Manual for instructions.

Cross-linking: CUT&Tag is a native procedure and works best for unfixed samples. This is an important advantage over ChIP-seq, which requires heavy cross-linking to stabilize targets on chromatin. Cross-linking is a major source of background and data variability in ChIP-seq, and contributes to low yields. Bypassing this step helps streamline CUT&Tag workflows, increases library yields, and reduces background, which improves assay reliability and enables profiling from low cell numbers.
When to use cross-linking: There are some instances where light cross-linking can be useful in CUT&Tag, such as for stabilizing potentially labile PTMs (e.g. histone acetylation). Note that we always recommend trying native conditions first, or at least in parallel with cross-linked samples. See our cross-linking protocol for details.
Number of nuclei: EpiCypher recommends using ~100,000 nuclei per reaction, particularly when mapping new targets or using new cell types. For nuclei prep, harvest 100,000 cells per reaction plus 10% excess to account for sample loss.
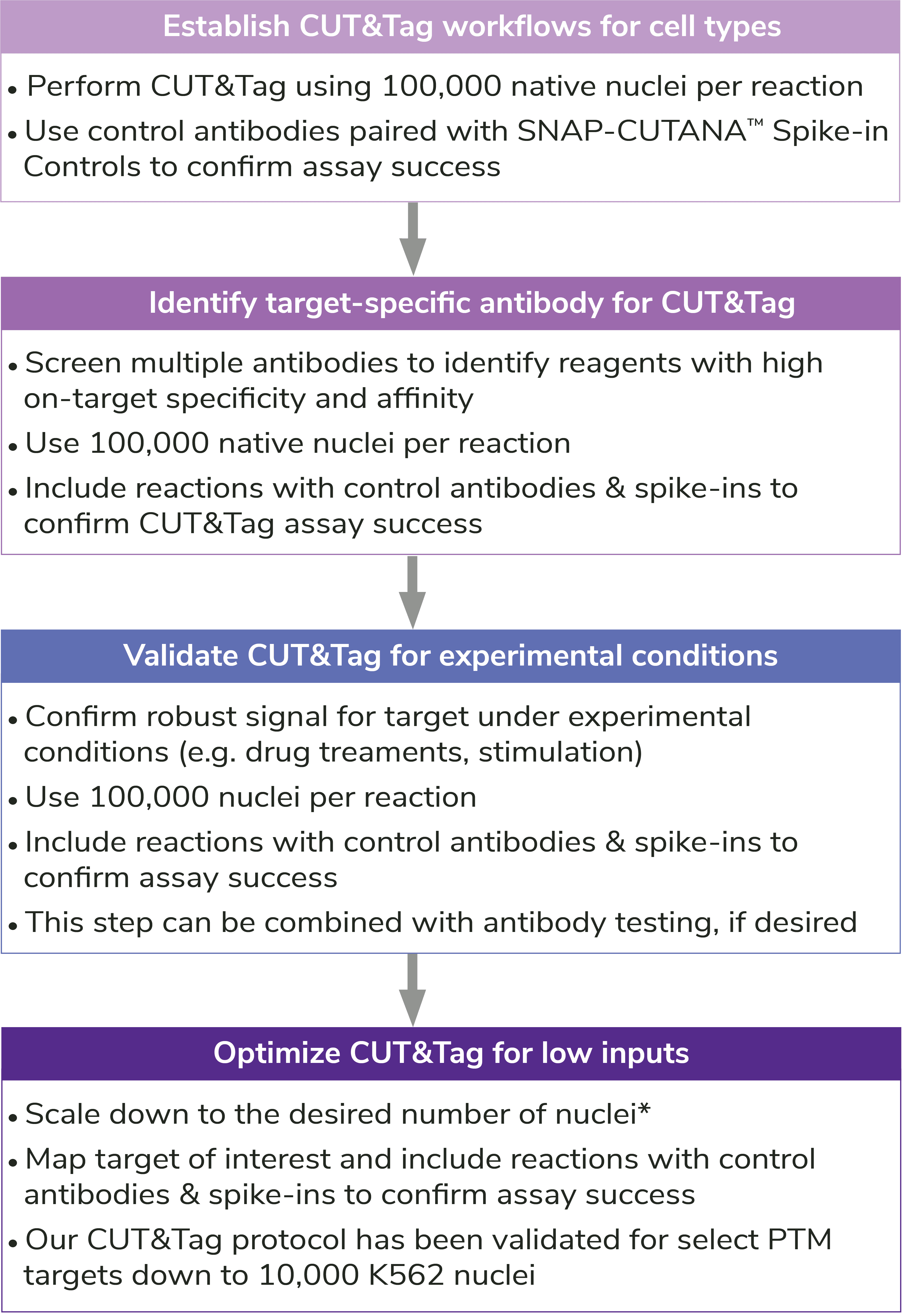
Reducing nuclear inputs: Following initial validation of CUT&Tag workflows using 100,000 nuclei and control antibodies (see Controls, below), scale down number of nuclei as outlined in Figure 5.
Replicates: CUT&Tag is reliable for histone PTM mapping. Two biological replicates (e.g. nuclei from two wild-type mice) per target are usually sufficient, although technical replicates may also be included.
II. Controls
Assay controls should be included in every CUT&Tag experiment
to confirm workflow success (Figures 4,5). EpiCypher offers
control antibodies and spike-in nucleosome controls to boost confidence in
CUT&Tag results.
Positive and negative control reactions: It is recommended to include reactions with negative (IgG, EpiCypher 13-0042) and positive (e.g. H3K27me3, EpiCypher 13-0055) control antibodies in every CUT&Tag experiment. Control antibodies should show reliable and accurate performance in CUT&Tag so they can be used to validate assay workflows. EpiCypher also recommends adding the SNAP-CUTANA™ K-MetStat Panel of nucleosome spike-in controls to these reactions.
Using control antibodies to validate workflows: EpiCypher always examines fragment distribution and library yields from control reactions (Figure 4). H3K27me3 positive control reactions should have an average fragment size of ~300 bp (see examples in Figure 3). Yields from H3K27me3 reactions and experimental targets are typically much greater than the IgG negative control, although yields may vary by cell type and starting cell number.
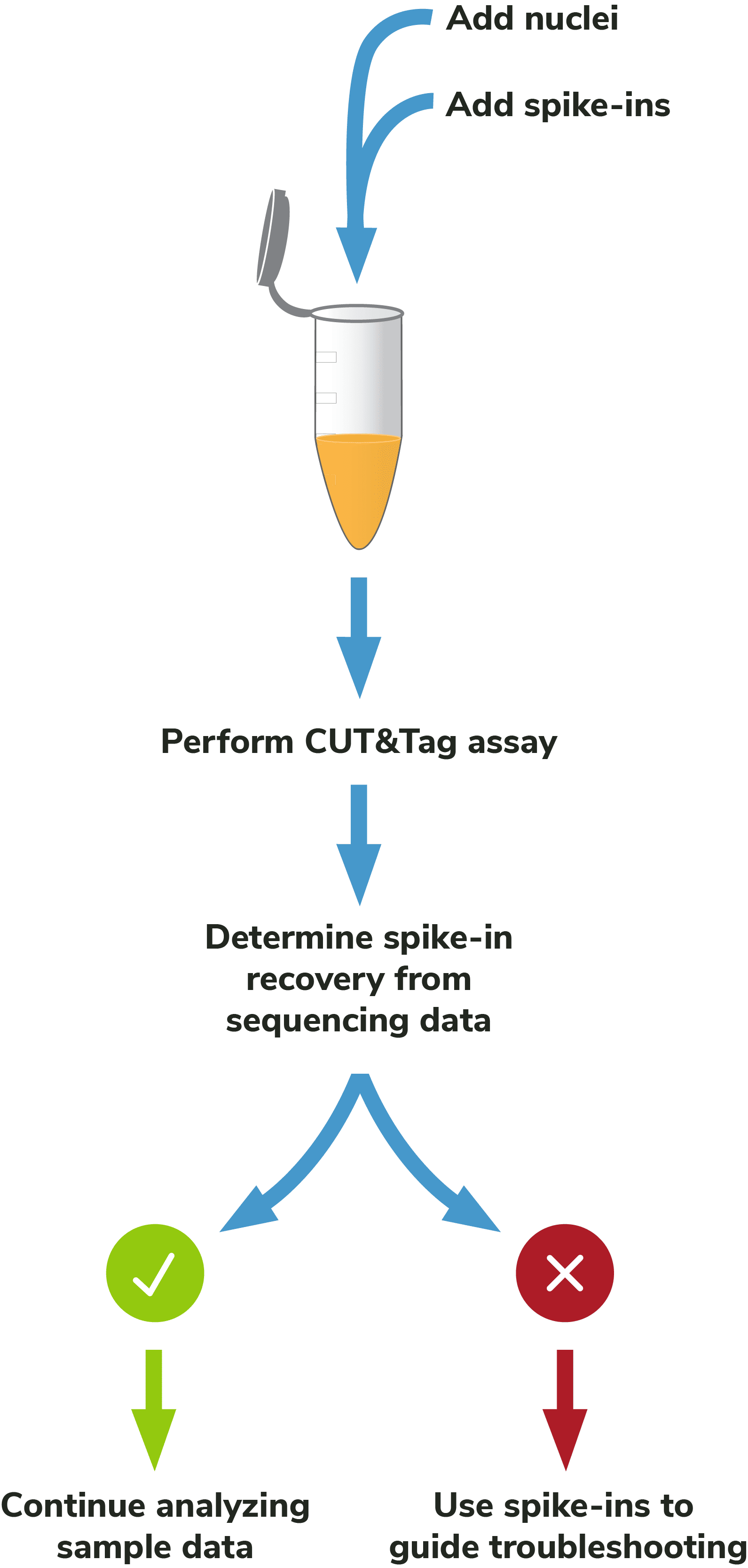
Adding spike-ins to determine assay success: To validate workflows and guide troubleshooting, EpiCypher recommends adding the SNAP-CUTANA™ K-MetStat Panel of nucleosome spike-ins to both positive and negative control reactions (Figure 5). Spike-ins are user-defined controls added to experimental samples that are used to gauge assay performance. The K-MetStat Panel is directly added to nuclei and processed alongside the sample throughout the CUT&Tag protocol (Figure 6). The spike-in data can be used to validate specific aspects of the workflow, such as antibody specificity or enzyme activity, and provide key insights for troubleshooting.
Although the SNAP-CUTANA K-MetStat Panel works like other genomic spike-ins, its design comes with added benefits. The K-MetStat Panel is the only control that uses purified recombinant nucleosomes, replicating the physiological target of CUT&Tag assays. The panel comprises 16 unique nucleosomes, each containing a distinct histone lysine methylation PTM (or unmodified) and DNA barcode, which provides both on- and off-target substrates for control reactions. As a result, spike-in data can directly report on antibody specificity, as well as pAG-Tn5 activity, sample quality, and more. For information on how to use the K-MetStat Panel for workflow validation and monitoring technical variability, see the SNAP-CUTANA™ User Guide (under Documents and Resources).
III. Target & antibody selection
Target compatibility: As mentioned above, CUT&Tag is most robust for histone PTMs. For chromatin-associated proteins, we generally recommend CUTANA™ CUT&RUN assays, which generate reliable profiles for diverse targets and sample types.
Antibody selection: CUT&Tag assays use a small number of cells to generate low yields with minimal background. Antibody performance is thus crucial for high-quality results. When selecting antibodies for CUT&Tag experiments, consider the following:
- Antibodies that work well in ChIP-seq are NOT guaranteed success in CUT&Tag.
- It is important to use a highly specific and efficient antibody that has been validated in CUT&Tag (if possible). Histone PTM antibodies are particularly susceptible to off-target binding and lot-variability, which can compromise biological interpretations16.
- If no CUT&Tag validated antibodies are available, it is recommended to test antibodies that have been validated for CUT&RUN. EpiCypher doesn’t currently lot-validate all antibodies for CUT&Tag applications, but many of our CUTANA SNAP-Certified Antibodies to histone PTMs can generate high-quality data by CUT&Tag.
How to validate antibodies: To identify the best antibody for your target, source 3-5 antibodies from various vendors that bind distinct target epitopes. Test side-by-side in CUT&Tag using 100,000 fresh, unfixed nuclei per reaction. Select a specific antibody based on DNA yield, enrichment, and signal-to-noise in sequencing data. Include positive and negative control reactions in every experiment to monitor variability and workflow success, and always perform quality checks to confirm sample prep (Figure 4). See Figure 5 for guidance.
Validating antibodies to histone lysine methylation PTMs. The SNAP-CUTANA K-MetStat Panel can be used to validate methyl-lysine PTM antibodies in CUT&Tag. Aim for less than 20% antibody cross-reactivity to off-target PTMs and consistent genomic enrichment with 100,000 nuclei.
Note on optimization for low numbers of nuclei: Antibodies validated at 100,000 nuclei may generate low yields or fail with <100,000 nuclei. This could be due to poor antibody binding efficiency and/or specificity, problems that are worsened when using fewer nuclei. A poor antibody also challenges profiling of low-abundance targets, e.g. H3K4me3. In these cases, it is recommended to screen additional target-specific antibodies CUT&Tag assays, as outlined in Figure 5. If no other antibodies are identified, it may be necessary to adjust the indexing PCR cycling conditions and increase sequencing depth (see more below).
Shop CUTANA™ CUT&RUN Antibodies
IV. Indexing PCR & sequencing
Indexing PCR: Use the minimum number of PCR amplification cycles that yield ~30 ng DNA (≥0.5 nM from Bioanalyzer/TapeStation results, 200-700 bp region). This strategy minimizes read duplicate rates while still providing sufficient library for accurate quantification and library pooling.
Sequencing: In general, 5-8 million sequencing reads are adequate for robust CUT&Tag profiles and will allow you to multiplex 10-100s of libraries per sequencing run. Paired-end sequencing (2 x 50 bp minimum) is recommended. Note that 5-8 million total sequencing reads per reaction should generate 3-5 million uniquely aligned reads.
Read duplicate rates are often high (up to 70%) due to increased assay sensitivity, but vary by target, cell type, antibody specificity, number of nuclei, and sequencing depth. However, even with high read duplication rates, robust tracks with good read diversity are obtained.
Troubleshooting low library yields: If library yields fall below 0.5 nM, it is recommended to repeat the assay using 100,000 nuclei per reaction, performing the quality control checks described above. If the library still has yields <0.5 nM, try increasing the number of indexing PCR cycles which will improve yields for fragment distribution analysis and enable pooling at standard concentrations for sequencing. Note that this strategy may increase read duplication rates.
Alternatively, if the experiment cannot be repeated, use a Speedvac to increase library concentration and add as much of the library as possible to the sequencing pool. In both cases, deeper sequencing is recommended to ensure sufficient read depth and fully capture library diversity.
Getting Started with CUT&Tag
Compared to ChIP-seq, CUT&Tag provides a streamlined and cost-effective approach for ultra-sensitive profiling of histone PTMs. The assay has been used to profile many cell and tissue types, including exhausted T cells17, rare muscle stem cells18, brain tissues13, clinical samples14,19, FACS sorted cells11, and plant tissues20, and is driving epigenomics towards new exploratory applications10,12,21.
Below we have compiled a list of our most popular CUT&Tag products, protocols, and blog posts to help jump start your CUT&Tag experiments. If you are profiling histone PTMs and need help deciding between CUT&RUN and CUT&Tag, stay tuned – that blog will be online soon!
- CUTANA CUT&Tag Products
- pAG-Tn5
- CUTANA CUT&Tag Kit
- H3K27me3 Positive Control Antibody
- IgG Negative Control Antibody
- SNAP-CUTANA K-MetStat Panel
- Anti-Rabbit Secondary Antibody
- Anti-Mouse Secondary Antibody
- CUT&Tag Protocols
- EpiCypher Blog Posts
Need more information about CUT&Tag?
Fill out form...
References
- Schmid, M. et al. ChIC and ChEC: Genomic Mapping of Chromatin Proteins. Mol. Cell 16, 147–157 (2004). PubMed PMID: 15469830.
- Skene PJ et al. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, (2017). PubMed PMID: 28079019.
- Skene, P.J. et al. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 13, 1006-1019 (2018). PubMed PMID: 29651053.
- Meers M.P. et al. Improved CUT&RUN chromatin profiling tools. eLife 8, e46314 (2019). PubMed PMID: 31232687.
- Kaya-Okur HS et al. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat Commun 10, 1930 (2019). PubMed PMID: 31036827.
- Kaya-Okur HS et al. Efficient low-cost chromatin profiling with CUT&Tag. Nat Protoc 15, 3264-83 (2020). PubMed PMID: 32913232.
- Henikoff S et al. Efficient chromatin accessibility mapping in situ by nucleosome-tethered tagmentation. Elife 9, (2020). PubMed PMID: 33191916.
- Wu SJ et al. Single-cell CUT&Tag analysis of chromatin modifications in differentiation and tumor progression. Nat Biotechnol (2021). PubMed PMID: 33846646.
- Janssens DH et al. CUT&Tag2for1: a modified method for simultaneous profiling of the accessible and silenced regulome in single cells. Genome Biol 23, 81 (2022). PubMed PMID: 35300717.
- Deng Y et al. Spatial-CUT&Tag: Spatially resolved chromatin modification profiling at the cellular level. Science 375, 681-6 (2022). PubMed PMID: 35143307.
- Bartosovic M et al. Single-cell CUT&Tag profiles histone modifications and transcription factors in complex tissues. Nat Biotechnol (2021). PubMed PMID: 33846645.
- Gopalan S et al. Simultaneous profiling of multiple chromatin proteins in the same cells. Mol Cell 81, 4736-46 e5 (2021). PubMed PMID: 34637755.
- Zhu C et al. Joint profiling of histone modifications and transcriptome in single cells from mouse brain. Nat Methods 18, 283-92 (2021). PubMed PMID: 33589836.
- Janssens DH et al. Automated CUT&Tag profiling of chromatin heterogeneity in mixed-lineage leukemia. Nat Genet 53, 1586-96 (2021). PubMed PMID: 34663924.
- Small EC et al. Chromatin Immunoprecipitation (ChIP) to Study DNA-Protein Interactions. Methods Mol Biol 2261, 323-43 (2021). PubMed PMID: 33420999.
- Shah RN et al. Examining the Roles of H3K4 Methylation States with Systematically Characterized Antibodies. Mol Cell 72, 162-77 e7 (2018). PubMed PMID: 30244833.
- Ford BR et al. Tumor microenvironmental signals reshape chromatin landscapes to limit the functional potential of exhausted T cells. Sci Immunol 7, eabj9123 (2022). PubMed PMID: 35930654.
- Robinson DCL et al. Negative elongation factor regulates muscle progenitor expansion for efficient myofiber repair and stem cell pool repopulation. Dev Cell 56, 1014-29 e7 (2021). PubMed PMID: 33735618.
- Amatullah H et al. Epigenetic reader SP140 loss of function drives Crohn's disease due to uncontrolled macrophage topoisomerases. Cell 185, 3232-47 e18 (2022). PubMed PMID: 35952671.
- Ouyang W et al. Rapid and Low-Input Profiling of Histone Marks in Plants Using Nucleus CUT&Tag. Front Plant Sci 12, 634679 (2021). PubMed PMID: 33912205.
- Sati S et al. HiCuT: An efficient and low input method to identify protein-directed chromatin interactions. PLoS Genet 18, e1010121 (2022). PubMed PMID: 35320278.